Применение данных фармакокинетики в клинике
Использование тестовой дозы. Если лекарство должно вводиться в/в, то изучение фармакокинетических параметров после однократного, относительно быстрого введения лекарства (болюса) в терапевтической дозе позволяет определить режим в/в поддерживающей инфузии либо повторных введений.
Поддержание постоянного фармакодинамического эффекта в соответствии с данными фармакокинетических исследований возможно при ряде условий. В случае в/в введения лекарства постоянство его концентрации в тканях будет зависеть от постоянства скорости инфузии. Время, необходимое для достижения плато концентрации, т. е. стационарного состояния (устойчивого динамического равновесия), определяется единственно константой скорости элиминации лекарства (kd) и не зависит от скорости инфузии лекарства.
При определенной скорости инфузии лекарства, если его период полувыведения выражается в минутах, то и плато концентрации будет достигнуто через время в минутах; если же период полувыведения выражается в часах, то и плато концентрации будет достигнуто через несколько часов.
В случаях длительного приема препарата внутрь средний стационарный уровень его концентрации в крови определяется величинами общего клиренса (Сl) и биодоступности (F). Чем выше Сl и чем ниже F, тем ниже будет его средняя концентрация в крови при данной схеме приема внутрь. Время достижения максимума концентрации (tмакс) и период пол у элиминации (t 1/2β) определяют размах колебаний уровня концентрации препарата в крови вокруг среднего стационарного уровня. Чем короче t 1/2β и tмакс, тем значительнее будет размах колебаний концентраций.
Правильная схема назначения лекарства внутрь должна учитывать период его полувыведения. Наиболее рационально первоначально назначать большую терапевтическую дозу лекарства с учетом массы тела и состояния больного, а далее — половину этой дозы в интервалы, равные периоду его полувыведения.
Например, если терапевтический эффект лекарства достигается при дозе 250 мг, а период полувыведения составляет 8 ч, то предпочтительно начать лечение с 500 мг, а далее — через каждые 8 ч по 250 мг. Чем меньше период полувыведения принятого внутрь лекарства, тем быстрее его концентрация в крови достигает уровня плато. Период полувыведения лекарства (t 1/2) в общем виде определяют по формуле, используя данные фармакокинетического исследования лекарства после однократной дозы:
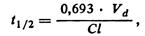
где Vd—кажущийся объем распределения лекарства в теле; Сl—клиренс лекарства.
При приеме внутрь схему индивидуального назначения лекарства можно определить по уравнению, предложенному Wagner J. G. и соавт. (1977):
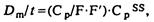
Где Ср—концентрация в плазме во время t; Dm — поддерживающая доза; t—одинаковый интервал между дозами; Dm/t—скорость дозирования; F—часть принятого лекарства, которая абсорбировалась; F'—часть абсорбированного лекарства, которая достигла системного кровообращения после эффекта «первого прохождения» через печень; тогда F*F’ — фактор биоусвояемости (биодоступности) лекарства, например, для таблеток дигоксина 0,6; Ср ss — концентрация лекарства в крови при стационарном состоянии (устойчивого динамического равновесия). Определить Cp/FxF' можно по уравнению:
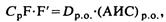
где Dp.o. — доза лекарства, принятого внутрь; (АИС)р.о. — общая площадь под кривой концентрация — время от 0 до определенного времени после одной дозы или же время под кривой в течение интервала между дозами при постоянной концентрации.
Этот же показатель (АИС) может экстраполироваться на неопределенное время. Он определяется как интервал уровня лекарства в крови (плазме) от 0 до неопределенного времени и измеряется количеством абсорбированного и находящегося в организме лекарства.
Суточную дозу препарата можно определить более простым методом, зная только значение общего (кажущегося) клиренса (CI, л/мин) и желаемую постоянную терапевтическую концентрацию препарата в крови (Css, мг/л или мкг/мл):
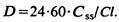
Однако эта формула не дает возможности определить частоту приема препарата, которая зависит от периода полувыведения препарата из крови (t 1/2β). Чем короче период t 1/2β тем чаще должен больной принимать препарат при той же суточной дозе.
В противном случае будут наблюдаться значительные колебания концентрации препарата в крови. При выборе частоты приема препарата в течение суток для поддержания постоянной концентрации препарата в крови можно пользоваться следующим правилом. Препараты с t 1/2β короче 4 ч принимать не менее 4 раз в день; при t 1/2β от 4 до 8 ч — не менее 3 раз в день, а при t 1/2β более 8 ч — 2 раза в день. Это правило не приемлемо для пролонгированных форм нитратов, так как длительность пребывания их в крови определяется не t 1/2β, а продолжительностью поступления лекарств в кровоток из лекарственной формы.
Соотношение эффективной концентрации препарата при внутривенном введении и приеме внутрь определяется для многих препаратов. Однако в ряде случаев эффективная концентрация в крови при быстром в/в введении препарата (например, верапамила) не предсказывает эквивалентную эффективную концентрацию в крови не только при приеме внутрь, но даже при медленной в/в инфузии. Это, возможно, связано с особенностями метаболизма и фармакологических эффектов отдельных стереоизомеров верапамила либо с появлением предполагаемых антагонистов — метаболитов при приеме внутрь или медленной в/в инфузии.
Определение индивидуальной особенности ф-кинетики лекарства у больного позволяет установить связь неэффективности лечения или его токсичности с концентрацией лекарства в крови. Поддержание постоянной концентрации лекарства в крови на терапевтическом уровне повышает эффективность лечения.
Контроль за постоянной концентрацией лекарства в крови затруднен при наличии нелинейной фармакокинетической характеристики, т. е. когда ф-кинетику лекарства нельзя описать как процесс первого порядка; изменение в дозе в этом случае будет вызывать большее, чем это должно было быть при линейной зависимости, повышение уровня лекарства в крови.
Использование ЭВМ позволяет ускорить процесс расчета фармакокинетических показателей. С помощью ЭВМ можно определить фармакокинетические параметры не только после тестовой однократной дозы лекарства, но и в течение повторных введений лекарства при стационарном состоянии концентрации препарата у больного, а также рекомендуемый режим введения лекарства по одному анализу крови (взятому в конце первого интервала между принятыми дозами) и по определению в нем концентрации лекарства.
Взаимоотношения постоянной концентрации лекарства в крови (стационарного состояния) и терапевтического эффекта являются сложными и неоднозначными. Следует иметь в виду возможный фармакологический эффект метаболитов лекарства и изменения в распределении его в крови. Следовательно, при выборе схемы и дозировки лекарств при наличии возможных активных метаболитов необходимо наряду с фармакокинетическими параметрами учитывать также фармакодинамические данные, а также возможность развития побочных явлений.
Косвенные методы определения фармакокинетических параметров лекарства. Если лекарство метаболизируется в основном в печени, то его клиренс может коррелировать с клиренсом эталонного (тестового) препарата (например, антипирина). В этих случаях фармакокинетические параметры можно косвенно и сравнительно просто определить по следующей пробе с антипирином. Больной принимает 1 г антипирина, растворенного в 250 мл воды, через 12 ч после приема пищи. Кровь (возможно и слюна) берут до и после приема антипирина. Клиренс антипирина подсчитывают и соотносят на единицу объема печени, определяемого методом ультразвукового сканирования.
Определение фенотипа ацетилирования как генетически детерминированного по активности N-ацетил-трансферазы позволяет классифицировать больных на «медленные», «быстрые» и «промежуточные» ацетилаторы. Это имеет значение для таких препаратов, как новокаинамид, гидралазин, салицилазосульфапиридин и др., для которых характерен метаболизм через ацетилирование. При исследовании больному дают внутрь, например, сульфадимезин (сульфаметазон) из расчета 10 мг/кг. Индекс ацетилирования в плазме (ИАП, %) наиболее информативен через 6 ч.
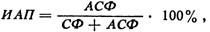
Где АСФ — концентрация ацетилированного, а СФ — неацетилированного препарата в крови.
У «быстрых» ацетилаторов быстро накапливаются токсические метаболиты, например, при приеме новокаинамида — N- ацетилпрокаинамид, вызывающий, в частности, такие токсические реакции, как синдром типа красной волчанки.
Основная задача фармакокинетических исследований сводится к выработке обоснованных рекомендаций по оптимальным дозировкам препаратов, обеспечивающим достижение достаточного терапевтического эффекта при отсутствии или минимальной частоте побочных явлений. Это особенно сложно и актуально при длительном медикаментозном лечении.
Например, мало еще известно об особенностях ф-кинетики на различных этапах длительного лечения, возможностях фармакокинетического взаимодействия при длительном комбинированном лечении, в частности: а) на стадиях абсорбции, метаболизма (индукция и ингибирование ферментных систем печени), б) при белковом связывании (механизм конкуренции), в) влияние на экскрецию неизмененного препарата и/или его метаболитов. Ряд этих актуальных вопросов изучается в исследованиях по профилактической фармакологии.
Дата добавления: 2023-08-09; просмотров: 769;
Поиск по сайту
Узнать еще
- Графические способы изображения статистических данных
- Двойной нож, рубанки и фуганки. Описание и применение
- Деятельность мозга – нормального и эпилептического. Система передачи и интерпретации данных сознания
- Искривление линий. Применение диафрагм
- Особенности оценки фармакодинамики сердечно-сосудистых лекарственных средств в клинике
- Применение автоматики в системах управления
- Применение биткоин-скриптов. Эффективные микроплатежи

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
