Клиническая характеристика хромосомных болезней
К хромосомным болезням относят группу врожденных патологий, которые возникают в результате нарушения числа и структуры хромосом в соматических и половых клетках человека. Общая популяционная частота этих аномалий около 1%. Как правило, это спорадические случаи, большинство хромосомных заболеваний (90%) возникает за счет новых мутаций. Исключение составляют транслокационные варианты, которые являются результатом сбалансированных транслокаций родителей.
4.5.1.Аутосомные синдромы
Известно, что среди живорожденных с аутосомными синдромами чаще всего встречаются полные трисомии по 13, 18 и 21 хромосомам, среди которых 75% приходится на долю синдрома Дауна.
Синдром Дауна
Первое клиническое описание этой аномалии относится к 1866 году и принадлежит английскому врачу Ленгтону Дауну. Спустя почти 100 лет цитогенетическую природу синдрома Дауна установил французский исследователь Ж. Лежен в 1959 году, обнаружив у больных лишнюю 21 хромосому. Еще до открытия Ж. Лежена в 1932 году Ваарденбург предположил, что причина болезни Дауна, возможно, связана с аномалиями хромосом. К настоящему времени болезнь Дауна изучена достаточно полно, так как представляет собой одну из самых частых хромосомных болезней (встречается с частотой 1:700 – 1:800). Среди всех умственно отсталых детей больные с синдромом Дауна составляют 10 – 12%. Соотношение полов при этом заболевании 1:1. На частоту рождения больных с синдромом Дауна не влияют расовые, географические и популяционные различия при сравнении одинакового возраста родителей. Частота рождения детей с синдромом Дауна зависит от возраста матери и в меньшей мере от возраста отца.
Причиной возникновения болезни Дауна является простое нерасхождение хромосом в мейозе. Вклад материнского нерасхождения составляет от 80 до 90%, а отцовского от 10 до 20%. Чем старше мать, тем больше риск появления ребенка с синдромом Дауна. Если возраст матери достигает 35 – 46 лет, то вероятность рождения больного ребенка может вырасти до 4%. Вероятность повторного возникновения синдрома Дауна в семье, где родители имеют нормальные кариотипы, не превышает 1 – 2%.
Цитогенетически болезнь Дауна представлена 3 формами:
· простая (регулярная) трисомия по 21 хромосоме (94 – 95% случаев);
· транслокация хромосомы 21 обычно на хромосомы группы D и G (3 – 4%);
· мозаицизм (1 – 2%).
Большая часть транслокаций при данном заболевании возникают за счет мутаций de novo. Одна четверть всех случаев транслокаций носят семейный характер, при этом повторный риск достигает 15% и во многом зависит от типа транслокации и от того, кто из родителей несет симметричную перестройку. Если же наследуемая транслокация представлена сочетанием двух хромосом 21q21, то повторный риск рождения больного ребенка 100%.
При молекулярно-генетических исследованиях удалось обнаружить критический район хромосомы 21, который, по мнению многих исследователей, несет ответственность за фенотипические проявления болезни Дауна. Полагают, что основную роль в возникновении умственной отсталости при этом заболевании играет увеличенная доза гена фермента супероксиддисмутазы, находящегося в районе длинного плеча хромосомы 21 (21q22).
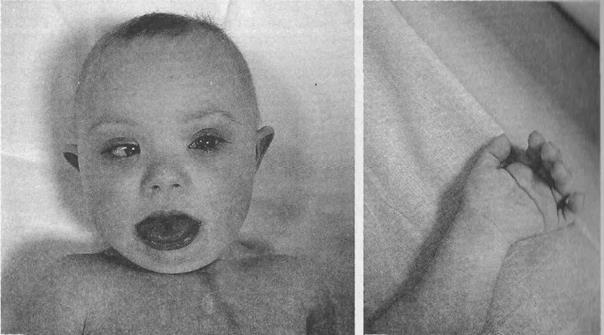
Больные с синдромом Дауна обычно невысокого роста, отличаются слабоумием и многочисленными физическими пороками. Они имеют характерную внешность и во многом очень похожи друг на друга. Диагностика этой болезни не представляет для акушера и педиатра особых затруднений даже у больных различных этнических групп.
4.5.1
Характерные признаки: мышечная гипотония, уплощенное лицо, монголоидный разрез глаз, эпикант, брахицефалия, короткий нос с широкой плоской переносицей, маленькие деформированные уши, полуоткрытый рот с высунутым утолщенным бороздачатым языком. Отмечаются катаракты, пятна Брушвильда (очаги белого цвета на границе наружной и средней трети радужки), косоглазие, разболтанность суставов.
При дерматоглифическом исследовании часто обнаруживается длинная поперечная складка на ладони (так называемая «обезьянья борозда»). В общей популяции этот признак встречается приблизительно у 1%, в то время как при синдроме Дауна его частота достигает 40%. Кроме того, у больных на мизинце имеется всего одна единственная складка (20 – 25%), которая довольно часто бывает симметричной на обеих руках.
Особенно часто у детей с болезнью Дауна наблюдаются пороки сердечно-сосудистой системы: дефект межжелудочковой перегородки, тетрада Фалло или незаращение артериального протока; иногда отмечаются пороки желудочно-кишечного тракта; гораздо реже встречаются пороки развития почек и мочевыводящих путей.
У больных с синдромом Дауна чаще возникают инфекционные и злокачественные заболевания, что, по-видимому, связано с нестабильностью и слабостью иммунной системы при этом заболевании.
При паталогоанатомическом исследовании размер и масса мозга больных из-за недоразвития уменьшены, ствол мозга и мозжечок маленькие, борозды и извилины развиты не полностью.
Одним из самых важных симптомов при синдроме Дауна является общее психическое недоразвитие. Олигофрения наблюдается от легких до тяжелых форм. Чаще всего у больных встречается имбецильность (65 – 90%), дебильность и идиотия диагностируются примерно в равном соотношении. На 1-м году жизни дети с таким заболеванием заметно отстают в моторном и психическом развитии. Они позже начинают сидеть и ходить, их мышцы резко гипотоничны, объем движений в суставах увеличен. Обучение во вспомогательных школах возможно, но не всегда; мыслительные процессы заторможены, читают и пишут с трудом, пересказывают только по вопросам, самостоятельный пересказ вызывает у них большие затруднения. Проведенные в последнее время исследования при болезни Дауна показали, что у них наблюдается более раннее развитие болезни Альцгеймера.
За последнее время продолжительность жизни больных с синдромом Дауна значительно увеличилась. Если раньше такие больные умирали в раннем детстве от различных инфекционных болезней, то теперь они доживают до 30 лет и более. Снижение продолжительности жизни, в основном, связано со снижением клеточного и гуморального иммунитета; у них нарушаются процессы репарации ДНК; они подвержены преждевременному старению, смерть часто наступает от сердечно-сосудистой недостаточности, инфекций, ряда онкологических заболеваний.
Лечение болезни Дауна малоэффективно, в основном оно симптоматическое. Широко применяются стимулирующая терапия. Медико-психологические, медико-педагогические и лечебные мероприятия позволяют адаптировать некоторых больных к посильной трудовой деятельности.
Диагноз болезни Дауна проводится на основании тщательного клинического обследования и обязательного цитогенетического анализа.
Если один из родителей является носителем сбалансированной транслокации с вовлечением 21 хромосомы, то при планировании деторождения в такой семье необходимо проводить дородовую диагностику, основанную на цитогенетическом и ультразвуковом обследовании плода. Кроме того, дородовую диагностику плода целесообразно проводить и у женщин старше 35 лет из-за повышенного риска рождения детей с синдромом Дауна.
Синдром Патау
Синдром Патау (синдром трисомии 13 хромосомы) впервые был описан в 1960 году. Частота встречаемости этого синдрома в популяции - 1:6000 - 1:13 000 рождений; соотношение полов 1:1. Как и при болезни Дауна, дети с синдромом Патау чаще рождаются у матерей старшего возраста; средний возраст матерей, родивших детей с трисомией 13 около 33 лет, отцов – 34 года. Цитогенетически этот синдром представлен двумя вариантами: простой трисомией и транслокационной формой. В основе синдрома Патау лежит нерасхождение хромосом в мейозе у одного из родителей (в основном у матери) по 13-й паре хромосом. В кариотипе больного наблюдается 47 хромосом с лишней хромосомой 13. Этот вариант встречается у больных с частотой от 80 до 85%; остальные 15 – 20% представлены транслокационными вариантами. При транслокационной форме в кариотипе больного имеется 46 хромосом. Уменьшение числа хромосом происходит чаще всего в результате слияния двух хромосом группы D или хромосом группы D и G. Реже обнаруживаются и другие цитогенетические варианты (изохромосома, мозаицизм и др. транслокации). Следует заметить, что средний возраст матерей, родивших детей с транслокацией хромосом D/D, не превышает 25 лет.
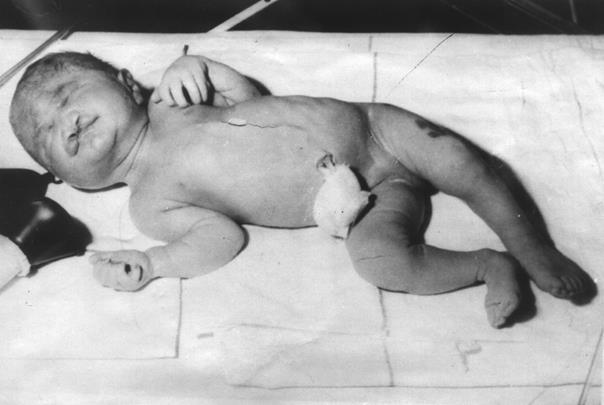
При рождении у детей с синдромом Патау отмечается пренатальная гипоплазия (масса тела не превышает 2,5 кг.); беременность осложняется многоводием (встречается до 50%).
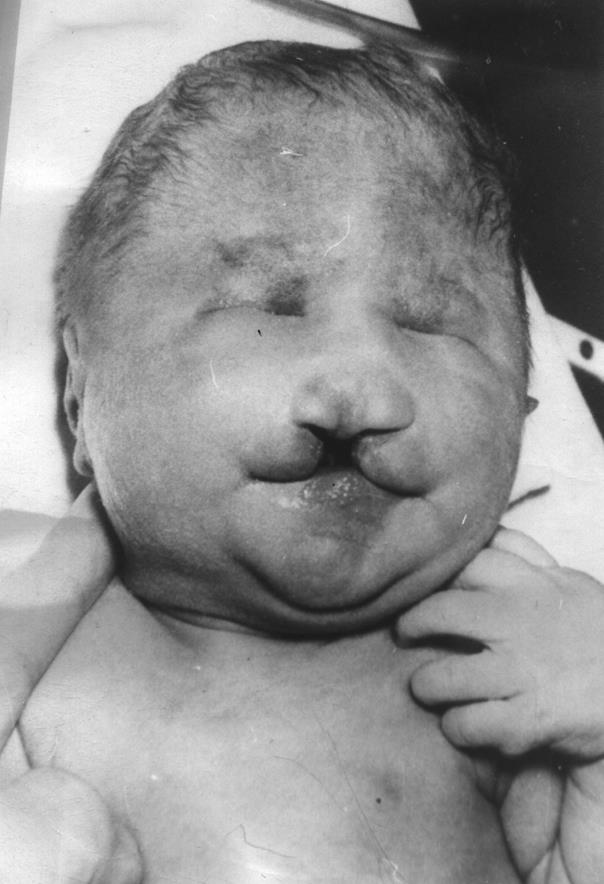
Внешний вид больных с синдромом Патау весьма специфичен. Клинически отмечается резкая умственная отсталость, выраженная микроцефалия, тригоноцефалия, неправильно сформированные и низко расположенные уши, аномалии глазного яблока (микрофтальмия и анофтальм), гипертелоризм, колобома радужки, помутнение хрусталика, одно- или двустороннее незаращение губы и неба, полидактилия, повышенная гибкость суставов, врожденные пороки внутренних органов (кардиоваскулярной и мочевой систем, желудочно-кишечного тракта), часто наблюдаются судороги. Из других клинических симптомов следует отметить гемангиомы на коже лица и рук, флексорную деформацию пальцев кисти, деформацию стопы, пупочные и пахово-мошоночные грыжи, крипторхизм, глухоту. Глухота у больных с трисомией 13 встречается в 80 – 85% случаев. Чаще всего изменения ограничены средним и нижней частью внутреннего уха.
4.5.2
4.5.3
При паталогоанатомическом исследовании бросаются в глаза множественные внешние и внутренние уродства практически всех органов и систем. Масса мозга уменьшена, часто отсутствует передний мозг; наблюдаются дефекты межжелудочковой и межпредсердной перегородок, камеры сердца расширены. Отмечается аномалия почек, мочеточников.
Из всех перечисленных аномалий ведущими, основными признаками синдрома Патау являются расщелина верхней губы и неба, полидактилия (часто двусторонняя) и глубокие поражения центральной нервной системы; в ряде случаев отмечаются достаточно грубые пороки – циклопия, этмоцефалия, цебоцефалия и др.
На основании клинических, дерматоглифических и паталогоанатомических данных диагноз поставить несложно. Окончательно он подтверждается цитогенетически. Дифференциальную диагностику следует проводить с врожденными пороками развития (синдромы Меккеля, Мора, тригоноцефалия Опица). Следует отметить крайне важное для практического врача обстоятельство – трисомные и транслокационные формы синдрома Патау по клиническим признакам неотличимы друг от друга, поэтому цитогенетическое исследование у больных для дифференциальной диагностики этих форм обязательно. При транслокационном варианте трисомии 13 вероятность повторного рождения аномального потомства высока, а при трисомном варианте она, вероятно, не превышает аналогичных показателей при болезни Дауна (1 – 2%).
Прогноз при синдроме Патау неблагоприятен, продолжительность жизни редко превышает 1 год, дети умирают от тяжелых пороков развития, несовместимых с жизнью.
Успешных методов лечения нет.
Синдром Эдвардса
Синдром Эдвардса описан в 1960 году. Частота его среди новорожденных колеблется от 1 на 7000 до 1 на 10 000 детей; девочки поражаются в 3 раза чаще, чем мальчики. Так же, как и при синдроме Дауна и Патау, имеется четкая зависимость частоты рождаемости детей с этим синдромом от возраста матери, но эта зависимость менее выражена. Риск родить больного ребенка не превышает 0,8%. Цитогенетически синдром Эдвардса представлен простой трисомией хромосомы 18 (90%), в 10 % случаев наблюдается мозаицизм, который встречается значительно чаще у девочек, чем у мальчиков: вероятнее всего это связано с большей жизнестойкостью женского организма.
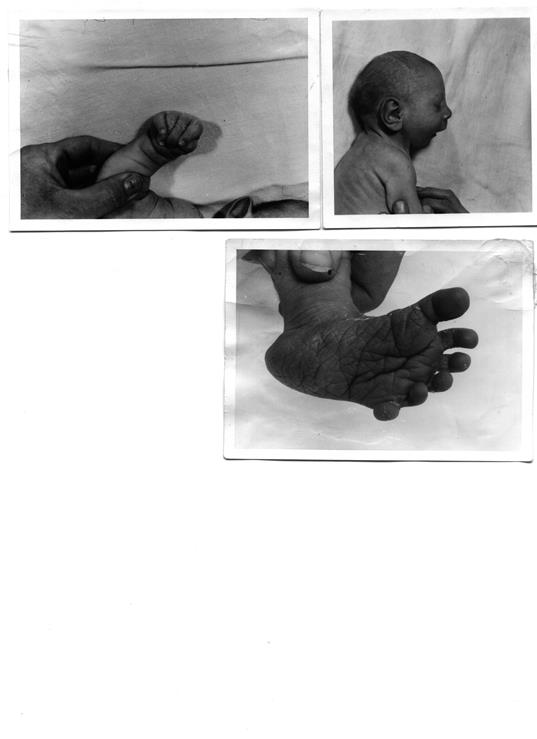
Больные дети часто рождаются недоношенными или переношенными, отмечаются слабая активность плода, многоводие. Дети часто рождаются в асфиксии, с низкой массой тела (2200 – 2400) и резкой гипотрофией. Череп маленький, сбоку сдавлен, затылочная часть вытянута, лоб маленький, уши расположены низко и их форма почти всегда аномальная, глазные щели узкие, наблюдаются гипертелоризм, эпикант, птоз, часты колобомы, микрофтальмия, катаракта, рот маленький, высокое небо, иногда с расщелиной. Шея короткая, иногда с крыловидной складкой, короткая грудная клетка, сердечный горб. Характерно расположение пальцев кистей – они согнуты. Второй палец перекрывает третий, остальные искривлены. Типична форма стопы в виде «качалки» (80%), часто наблюдается косолапость. Постоянны пороки сердца, почек, пищеварительного тракта. У 100% больных отмечается сниженный интеллект, часто – идиотия и имбецильность, реже дебильность. Во всех случаях наблюдается нарушение развития головного мозга.
4.5.4
Дерматоглифическая картина типична: на кончиках пальцев рук преобладают дуги или плоские петли (реже), в результате чего общий гребневой счет чрезвычайно низкий. Часто наблюдается поперечная складка ладони.
Цитогенетически у 80% больных обнаруживаются трисомия по хромосоме 18, у 10% - мозаицизм, в остальных случаях имеются другие хромосомные нарушения.
Дифференциальная диагностика очень сложна.
Цитогенетическое исследование должно проводиться во всех случаях для подтверждения диагноза и определения риска рождения будущего потомства.
Клинические проявления при синдроме Эдвардса гораздо тяжелее, чем при синдроме Дауна.
Продолжительность жизни чаще не более 6 месяцев, лишь 50% детей доживают до 2-х месячного возраста, около 10% живут 1 год; некоторые дети доживают до 10 лет. Причина смерти – сердечная недостаточность или инфекционные заболевания.
Дата добавления: 2016-10-26; просмотров: 3239;
Поиск по сайту
Узнать еще
- Cравнительная характеристика усилителей на БТ
- I. Загальна характеристика
- II. Монозы и их характеристика
- Nemathelmintes. Общая характеристика типа. Nematoda. Характеристика класса. Медицинское значение. Био- и геогельминты.
- Plahelmintes (Плоские черви).Общая характеристика типа. Морфология, систематика, основные представители, значение.
- Protozoa. Общая характеристика подцарства. Классификация. Представители. Медицинское значение.
- V. Механическая характеристика
- V. Упрощенная схема замещения трансформатора и внешняя характеристика.

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
