Синдром Шерешевского-Тернера.
Впервые клиническую картину данного синдрома описал Шерешевский Н.А. в 1925 году. Классическое описание принадлежит Тернеру Х. Х. (1938). Цитогенетическую природу заболевания открыл Форд С.Е. в 1959 году, обнаружив кариотип 45, ХО.
Это единственная форма моносомии, обнаруженная у человека. Частота встречаемости синдрома ХО по разным источникам колеблется от 1 на 1000 до 1 на 7000 и более. Такое разночтение частоты встречаемости данного синдрома может быть объяснено не только присутствием в кариотипе мозаичных вариантов, но и тем, что при различных структурных перестройках Х-хромосомы (изохромосомы, делеции короткого и длинного плеча, кольцевые хромосомы, Х-транслокации) наблюдается одна и та же клиническая картина. По последним уточненным данным моносомия по Х хромосоме встречается с частотой от 0,1 до 0,4 на 1000. Синдром Шерешевского-Тернера обнаруживается приблизительно при 1% всех зачатий, среди спонтанных абортусов его находят в 19% случаев; 95% зигот с хромосомным набором погибает внутриутробно.
Кариотип 45, ХО характеризуется большой цитогенетической и клинической вариабельностью. Приблизительно у 60% больных в кариотипе содержится только одна Х-хромосома, в остальных случаях наблюдаются различные типы структурных и числовых нарушений Х-хромосомы. В 80 - 85% случаев единственная Х-хромосома имеет материнское происхождение и лишь в 15 -20% - отцовское.
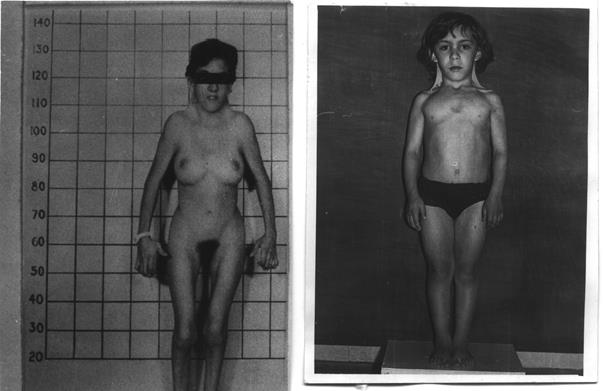
Клинические симптомы заболевания проявляются с первых дней жизни. Масса тела детей при рождении снижена, отмечается лимфатический отек верхних и нижних конечностей, низкий рост волос на шее. Отек стоп и голеней может держаться от 2 до 3 лет. В течение 1 года жизни ребенок постепенно отстает в росте, особенно заметно замедление роста в 9 – 10 лет. В дальнейшем для таких больных - низкий рост – является одним из самых характерных признаков; у взрослых он не превышает 140 - 145 см.
Для больных с синдромом Шерешевского-Тернера характерны кожные крыловидные складки на короткой шее (до 60%), широкая грудная клетка (60%), Х-образное искривление голеней (56%). При полной форме синдрома Шерешевского-Тернера наблюдается половой инфантилизм, первичная аменорея и бесплодие (90%), внешние и внутренние половы органы недоразвиты, отсутствуют матка и фаллопиевые трубы; наблюдается недоразвитие вторичных половых признаков, связанное с недостатком эстрогенов, которые приводят к недоразвитию молочных желез и скудному оволосению на лобке и в подмышечных впадинах. Поражаются сердечно-сосудистая, мочеполовая, скелетная и кожные системы. При дерматоглифическом обследовании отмечаются дистально расположенный осевой трирадиус, поперечная ладонная складка, увеличение частоты узоров в области гипотенара и высокий гребневой счет. Интеллектуальное развитие нормальное или близкое к норме.
4.5.7
При патологоанатомическом исследовании вместо гонад у таких больных находят недифференцированный тяж, не содержащий фолликулов и секреторных клеток. В 60% случаев встречаются аномалии мочевой системы, чаще подковообразная почка, удвоение почек и мочевыводящих путей; реже описывают врожденные аномалии сердца (20%).
Предварительный диагноз синдрома Шерешевского-Тернера основан на характерной клинической картине и исследовании полового хроматина, окончательный – на результатах цитогенетического анализа и применении высокоразрешающих молекулярно-цитогенетических методов. Последние методы необходимо применять в случаях определения происхождения маркерных хромосом (минихромосомы) и низкого содержания мозаичных клеток в кариотипе (до 20%).
Дифференциальную диагностику проводят с синдромом Боневи-Ульриха - аутосомно-доминантной болезнью, при которой у некоторых больных сохраняется генеративная функция, наблюдается передача патологического гена или генов из поколения в поколение и отсутствует характерная цитогенетическая картина (ХО). Кроме того, синдром ХО необходимо отличать от синдрома Нунан, смешанной дисгенезии гонад, чистой дисгенезии гонад 46, ХХ и чистой дисгенезии гонад 46, ХУ
Лечение в основном симптоматическое и обычно направлено на коррекцию вторичных половых признаков. Лечебные мероприятия проводят обычно эндокринологи (эстрогены, гормон роста), пластические хирурги (удаление крыловидных складок), психотерапевты; при стертых мозаичных формах синдрома показана гормональная заместительная терапия.
Синдром трипло-Х
Впервые синдром трисомии по Х-хромосоме был описан П. Джекобс с соавторами в 1959 году.
Они обнаружили в ядрах эпителия слизистой оболочки щеки больной два тельца полового хроматина. В среднем женщины с кариотипом ХХХ встречаются с частотой 1 - 1,4 на 1000 родившихся девочек. Кроме обычного трисомного варианта 47, ХХХ у женщин описаны полисомии по Х-хромосоме с кариотипами 48, ХХХХ и 49, ХХХХХ.
Клиническая картина этого заболевания чрезвычайно разнообразна. Психиатр, эндокринолог и гинеколог могут встретиться как с отчетливыми клиническими проявлениями этого синдрома, так и со стертыми формами. Всем больным свойственно только присутствие в кариотипе трех хромосом Х. Около 30% таких больных сохраняют генеративную функцию и имеют нормальных детей.
Клинически больные с трипло-Х имеют недоразвитые яичники, гипоплазию матки, нерегулярный менструальный цикл; у них рано наступает вторичная аменорея или бывает преждевременный климакс. У многих больных обнаруживаются неспецифические соматические дизморфии различной выраженности; грубых аномалий развития наружных половых органов не обнаружено. Довольно часто у женщин с ХХХ-хромосомным комплексом отмечается незначительное снижение интеллекта в стадии дебильности. Доказано, что среди них в несколько раз чаще можно встретить лиц с психопатическими чертами и наклонностью к расстройствам шизофреноподобного круга. По данным Филлипова Ю.И. (1971) у взрослых больных с синдромом трипло-Х шизофрения протекает неблагоприятно с выраженными изменениями личности; они склонны к проявлению эпилепсии, особенно в детском возрасте. У таких больных частота трипло-Х в несколько раз выше популяционных показателей. На цитогенетическое исследование больные чаще всего попадают из психиатрических лечебниц и домов инвалидов для детей, которые страдают умственной отсталостью.
Многие исследователи отмечают своеобразную особенность: с увеличением числа Х- хромосом в кариотипе до 4, 5 и более клинические проявления синдрома усиливаются. Больные, имеющие 4, 5 или более Х-хромосом, умственны более отсталые и, как правило, из-за эндокринного дисбаланса у них резко нарушается генеративная функция.
Предварительный диагноз синдрома трипло-Х основан на исследовании полового хроматина, Этот метод позволяет различать больных с аномальным комплексом Х-хромосом и первичной эндокринной патологией. Окончательный диагноз устанавливается по результатам кариологического исследования.
Больные с трипло-Х могут иметь потомство.
Лечение в основном симптоматическое и направлено на коррекцию эндокринного дисбаланса, в первую очередь на устранение нарушений функции яичников.
Дата добавления: 2016-10-26; просмотров: 3632;
Поиск по сайту
Узнать еще
- I. РАЗДЕЛ ПО ПРОБЛЕМЕ НЕДОСТАТОЧНОСТИ МИТРАЛЬНОГО КЛАПАНА (СИНДРОМ МИТРАЛЬНОЙ РЕГУРГИТАЦИИ)
- II. РАЗДЕЛ ПО ПРОБЛЕМЕ СТЕНОЗА МИТРАЛЬНОГО ОТВЕРСТИЯ ( СИНДРОМ МИТРАЛЬНОЙ ОБСТРУКЦИИ ).
- А Нейропсихологические синдромы поражения задних отделов коры больших полушарий головного мозга.
- Абеталипопротеинемия (синдром Бассена-Корнцвейга)
- Абстинентный синдром
- Альтернирующие синдромы при поражении ствола головного мозга
- АНТИФОСФОЛИПИДНЫЙ СИНДРОМ
- Антифосфолипидный синдром и волчаночный антикоагулянт

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
