Синдром «кошачьего крика»
Синдром «кошачьего крика» впервые описал Дж. Лежен с соавторами в 1963 у 3 детей с множественными аномалиями, глубокой умственной отсталостью и характерным плачем, который напоминал кошачий крик. Цитогенетически у всех больных обнаруживается укорочение приблизительно на треть и более короткого плеча одного из гомологов 5-й хромосомы. В коротком плече находится участок (15,1 – 15,2), который непосредственно вызывает развитие этого синдрома. Кроме обычной делеции, хромосомная перестройка может быть представлена другими вариантами (кольцевая хромосома, транслокация, мозаицизм по делеции). Около 85% всех случаев синдрома «кошачьего крика» являются спорадическими, 15% наследуются от фенотипически нормальных родителей - носителей сбалансированных перестроек. Этот синдром встречается гораздо чаще других синдромов, связанных с делециями аутосом; частота его примерно – 1 на 45 000.
Клинически синдром «кошачьего крика» очень полиморфен. Корреляцию между величиной делеции хромосомного материала и клиническими симптомами установить весьма трудно. Без своеобразного крика у больного надежный диагноз до цитогенетического исследования установить невозможно, так как большинство клинических симптомов этой болезни встречается и при других хромосомных аномалиях. В типичных случаях у детей с синдромом «кошачьего крика» клинически отмечают круглое лицо с гипертелоризмом, антимонголоидные глазные щели, косоглазие, эпикант, уменьшенный подбородок, плоскую спинку носа, деформированные и низко расположенные уши, короткую шею, нижнюю синдактилию, укороченные пальцы, клинодактилию, врожденные пороки сердца и половых органов, аномалии почек, атрофию зрительного нерва. С возрастом некоторые клинические признаки постепенно исчезают и среди них «кошачий крик», лунообразное лицо, мышечная гипотония. В то же время нарастает отставание умственного и физического развития, косоглазие, микроцефалия.
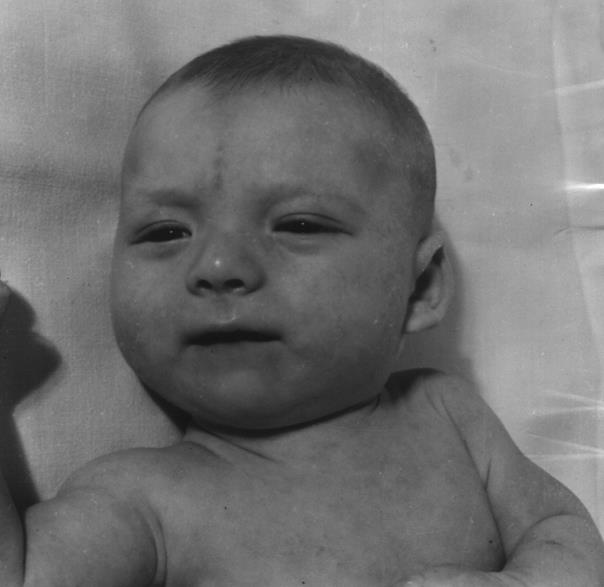
4.5.6.
При паталогоанатомическом исследовании находят микрогирию и гипоплазию мозжечка, уменьшенный мозг, расширенные желудочки мозга, гипоплазию лобных долей, аринэнцефалию, различные пороки сердца, аномалии почек, крипторхизм, экзофтальм, гемангиомы.
Продолжительность жизни больных с данным синдромом зависит от тяжести врожденных пороков развития; большинство из них умирает рано, некоторые доживают до 10-летнего возраста и более (около 14%).
Лечения нет (паллиативная терапия).
Во всех случаях для уточнения диагноза у больного и расчета риска прогноза потомства в семье показано цитогенетическое обследование, так как среди некоторых семей наблюдаются носители сбалансированных транслокаций.
В клинической практике встречаются и другие частичные анеуплоидии (трисомии, моносомии): 9р+; 11q+;18р-; 18q-; 21q-; 22q-; делеции коротких плеч акроцентрических хромосом (13 – 15; 21 – 22) практически не имеют каких-либо клинических проявлений. Более подробную информацию об этих и других хромосомных аномалиях можно узнать из монографии Ворсановой С.Г. и др., (2006).
Дата добавления: 2016-10-26; просмотров: 2305;
Поиск по сайту
Узнать еще
- I. РАЗДЕЛ ПО ПРОБЛЕМЕ НЕДОСТАТОЧНОСТИ МИТРАЛЬНОГО КЛАПАНА (СИНДРОМ МИТРАЛЬНОЙ РЕГУРГИТАЦИИ)
- II. РАЗДЕЛ ПО ПРОБЛЕМЕ СТЕНОЗА МИТРАЛЬНОГО ОТВЕРСТИЯ ( СИНДРОМ МИТРАЛЬНОЙ ОБСТРУКЦИИ ).
- А Нейропсихологические синдромы поражения задних отделов коры больших полушарий головного мозга.
- Абеталипопротеинемия (синдром Бассена-Корнцвейга)
- Абстинентный синдром
- Альтернирующие синдромы при поражении ствола головного мозга
- АНТИФОСФОЛИПИДНЫЙ СИНДРОМ
- Антифосфолипидный синдром и волчаночный антикоагулянт

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
