СПОСОБЫ АКТИВАЦИИ ХИМИЧЕСКОЙ РЕАКЦИИ
Этот раздел вряд ли можно отнести к фундаментальным разделам химии, но, в целом, приемы, которыми пользуются в лабораториях мира для проведения того или иного химического взаимодействия и тем более взаимодействия, которое не «желает идти по хорошему», безусловно базируются на фундаментальных знаниях.
Гораздо хуже в этом отношении обстоит дело с химическим материаловедением. Как правило, между веществом и материалом стоит или стоят одна или несколько стадий переработки, модифицирования или иного действия в основном физического свойства. Конечно, в ряде случаев этого физического «передела» или «доводки» вещества, приводящего к приобретению им полезного свойства избежать нельзя, но одной из задач современного химического материаловедения является разработка таких технологий, которые дали бы возможность получать нужный материал в рамках чисто химических или физико-химических процессов.
Большинство из таких известных процессов объединяются под пока условным называнием – процессы «мягкой химии» (soft chemistry). Нельзя сказать, что это удачное название, поскольку в русском варианте оно как бы подразумевает мягкие условия проведения химической реакции. В действительности условия могут быть самые разные, в том числе и достаточно жесткие. Суть заключается в направленности получения нужного вещества или материала в рамках химических процессов с использованием промежуточных фаз или соединений, специально приготовленных для получения нужной фазы, вещества, материала. Обычно этот прием позволяет заметно снизить температуру или время образования конечного продукта. Иногда это помогает остановить процесс на стадии синтеза метастабильных фаз.
В этом разделе рассмотрим некоторые типичные и не очень «типичные» в экспериментальной химии способы активации химических реакций, в том числе и те, которые направлены на синтез материалов.
Мы знаем три термодинамических параметра, которые определяют химический процесс - температуру, давление и концентрацию, хотя сейчас все более активно вводится в рассмотрение четвертый параметр – размер частицы, - и три среды, в которых проводятся химическая реакция – раствор, газ и твердая фазы. В качестве самостоятельных рядом авторов рассматриваются процессы, проводящиеся в условиях плазмы, суперкритических средах и с участием частиц наноразмерного диапазона, но это дело вкуса, политики и амбиций автора и принципиально не изменяет сложившуюся классификацию.
Реакции с участием жидкой фазы. Безусловно, наиболее распространены методики и химические технологии, в которых в качестве среды используется тот или иной тип растворителя. В связи с этим целесообразно рассмотреть важнейший вопрос в химии, связанный с выяснением роли растворителя в ходе осуществлении той или иной химической реакции или какого-либо физического процесса.
В соответствии с современными представлениями большая часть известных растворителей даже под именем «инертный» выполняют функцию кислоты или основания. В соответствии с своими свойствами растворители «классического» типа делят на два больших класса – протонные, молекулы которых уже имеют или потенциально могут иметь подвижный протон, и апротонные, не имеющие протонов с таковыми свойствами, хотя, в принципе, в их составе атомы водорода и могут быть. Несколько в стороне стоят новые типы растворителей – ионные жидкости, представляющие собой низкоплавкие соединения, состоящие из органических, неорганических веществ, эвтонические смеси веществ или чисто солевые расплавы (эвтектики), и отдельно флюиды.
Протонные растворители – являются полярными веществами - это вода, карбоновые кислоты, амиды кислот, жидкий аммиак и др.
Апротонные растворители, например простые эфиры, могут быть полярными или неполярными и довольно часто используются в синтетической органической и металлорганической химии.
В свою очередь эти два больших типа растворителей могут быть разделены на кислоты и основания, обычно в льюисовском смысле, которые в свою очередь могут быть поделены на полярные и аполярные.
По своим свойствам все растворители характеризуются диэлектрической проницаемостью (e) и дипольным моментом (m).
К аполярным апротонным растворителям обычно относят растворители с величинами e<15 и m<2. Это третичные амины, галоидные алкилы, некоторые углеводороды. Взаимодействие аполярных апротонных растворителей с растворенным веществом невелико и в основном обусловлено силами Ван-дер-Ваальса.
К полярным апротонным растворителям относят растворители с e>15 и m>2. Это сульфоксиды, нитрилы, формамиды и др. Полярные апротонные растворители, обладая хорошими основными свойствами, взаимодействуют с растворяемым веществом по донорно-акцепторному (например, хлористый алюминий–эфир) или ион-дипольному (иодистый литий-эфир) типу, сольватируя положительно заряженную частицу. Соответственно, анионы практически не сольватируются этими веществами, но достаточно хорошо сольватируются протонными полярными растворителями, содержащими OH и NH-группы способными к образованию водородных связей.
При растворении ионных солей в раствор переходят либо ионы, либо их ассоциаты, например ионные пары. При растворении ковалентных соединений в растворе, наряду с нейтральными молекулами, могут происходить процессы ионизации, диссоциации и еще более глубокий процесс - аутокомплексообразование. Эта глубина, прежде всего, зависит от свойств растворяемого вещества и свойств растворителя. Так, трифенилхлорметан в уксусной кислоте с e=6 за счет образования водородных связей и сольватации хлоридного аниона только ионизируется. Следующая стадия – стадия диссоциации, связанная с резким увеличением электропроводности раствора, требует растворителя с более высокими значениями e. Но при этом растворитель должен обладать и высокой сольватирующей способностью. Например, нитробензол с e=45 не способен к этому из-за слабого сольватирующего действия. В «хорошем» растворителе с e>40 степень диссоциации электролита составляет не менее 50%, и, естественно, что замена растворителя подчас приводит к радикальному изменению скорости конкретной реакции, в которой принимают участие вещества способные к ионизации и диссоциации.
Чисто качественная теория, объясняющая роль растворителя в протекании органических реакций, предложена К. Ингольдом и Е. Хьюзом в 1930 г. Она основана на рассмотрении моделей переходных состояний, возникающих в ходе реакции, и показывает, что, если в переходном состоянии возникают заряженные частицы, реакция ускоряется в полярном растворителе, так как в нем снижается ее энергия активации.
Как отмечалось выше, несколько в стороне от «типичных» растворителей, стоит новый класс веществ, частично выполняющих те же функции, что и обычные растворители, т.н. «ионные» жидкости, но их свойства и природа радикально отличаются от первых. (Это очень молодой класс веществ, выделившийся в самостоятельную группу всего 10-12 лет тому назад, и желающие ознакомится с ним более подробно могут это сделать, прочитав обзор в J. of Molecular Catalysis A, 2002, v. 182-183, p.419-437). Тем не менее, к ним применимы многие положения и прогнозы, сделанные для классических растворителей. Большинство из этих жидкостей получается в результате реакции «квартеризации» при взаимодействии алкилов аммония или фосфония с алкилсодержащими реагентами. При этом образуются алкиламмониевые или фосфониевые соли R4E+X-. Возможны и другие приемы ионизации компонентов, которые приводят к низкоплавким (<100 оС) веществам, например, использование пиридиниевого или имидазольного ионов, введение кислот Льюиса или кислот Бренстеда (протонирование), обмен ионов и т.д. При этом совсем не обязательно удовлетворения требования стехиометрического соотношения реагентов, что сближает рассматриваемый тип веществ с смесями, составленными при сечении бинарной или более сложной системы.
Почему исследователи обратили внимание на эти вещества? Во-первых, крайне низкое собственное давление пара, что позволяет легко и нацело отделять более летучие, например органические продукты реакции, ликвидировать все технологические «сдувки» и потери растворителей в циклах разделения, регенерации, перегонки и т.п. Далее следует отметить довольно высокая термическую устойчивость ряда ионных жидкостей. Возможность в широких пределах изменять их сольватирующую способность и, соответственно, растворимость субстратов, путем модификации состава «растворителя» за счет введения различных заместителей в катионную часть или меняя анион; водорастворимость некоторых жидкостей или, напротив, нерастворимость, но устойчивость к действию воды, что позволяет проводить реакции в двухфазных областях. Возможность изменения в очень широком диапазоне кислотности и координирующей способности в ряду соединений с одним и тем же катионом, но с разными анионами. Например [NRR’R’’R’’’]Cl с преимущественно основными и сильными координирующими свойствами через нейтральный [NRR’R’’R’’’]AlCl4, являющийся слабо координирующим веществом, к сильной и некоординирующей кислоте [NRR’R’’R’’’]Al2Cl7. Эти, безусловно, замечательные свойства позволили апологетам этого направления объявить ионные жидкости панацеей от всех химических проблем, возникающих в процессе химических превращений, и поместить их в титул журнала и соответствующих программ Green Chemistry с соответствующим названием “green solvents”. Конечно, это явный перебор, поскольку компоненты этих жидкостей и сами жидкости после реализации чудесных превращений и многократного использования все же надо как-то утилизировать, а судя по составам некоторых из них, например хлоралюминатов или фторидов сурьмы (комплексные анионы), это далеко не простая и отнюдь не «зеленая» задача.
Тем не менее, при осуществлении некоторых процессов ионные жидкости, как и другие не менее экзотические растворители - флюиды (см. ниже), оказываются весьма эффективными. Прежде всего, это касается ряда каталитических реакций, в которых ионные жидкости могут выступать и как растворители и как сокатализаторы в процессах с участием соединений переходных металлов как катализаторов. Например, реакции гидрирования алкенов родиевыми катализаторами в слабокоординирующей ионной жидкости на основе имидазола и фторидов бора, сурьмы или фосфора (противоионы). Реакции окисления в той же жидкости саленовыми комплексами Mn(III). Гидроформилирование, реакция Хека (наращивание углеродной цепи за счет реакции галогенидов аренов с сложными эфирами алкенов, например бутилакриалтом), гидродимеризация, теломеризация (например сшивка бутадиена в присутствии воды и Pd катализатора), олигомеризация и полимеризация, метатезис олефинов, биотехнологические процессы и обычные органические реакции, начиная с реакций Дильса-Адлера, реакции Виттига, бензоиновой конденсации и кончая реакциями Фриделя-Крафца и реакциями синтеза металлорганических соединений. В настоящее время число примеров применений ионных жидкостей в органическом, металлорганическом синтезе и каталитических реакций достигает сотен и расширяется очень быстрыми темпами.
Рассмотренные растворители, независимо от их свойств, являются жидкостями и, как правило, используются при атмосферном давлении. Но помимо них в настоящее время приобретают все большую популярность растворители принципиально иного типа, называемые суперкритическими жидкостями и существующие, как правило, при высоких и очень высоких давлениях. Суперкритическое состояние растворитель или в общем случае вещество достигает тогда, когда при строго определенных критических температуре и давлении, например для воды это 374.2 о и 217.6 атм, для СО2 31.1 о и 72.8 атм, а для аммиака 132.5 о и 111.3 атм, плотности жидкости и пара сравниваются. Вещество в этом состоянии называют флюидом и его свойства уже мало напоминают его свойства в исходном состоянии – это уже не жидкость, так как их свойства радикально отличны, но и не газ, так как плотность флюида существенно выше и он не подчиняется газовым законам. Так, свойства воды как растворителя и реагента меняются вследствие изменения ее диэлектрической проницаемости e (1-2 вместо 78), плотности r (100-150 вместо 1000 кг/м3), электропроводности, ионного произведения, количества и качества водородных связей и всех прочих физических и структурных свойств. Следует отметить, что эти свойства меняются не скачкообразно, а непрерывно по мере увеличения температуры и давления.
В настоящее время флюидное состояние объясняют наличием в нем свободных молекул и различных слабосвязанных кластеров или ассоциатов с расстояниями между ними существенно большими, чем в классических жидкостях, но меньшими, чем в газах. Так, результаты компьютерного моделирования указывают на то, что степень ассоциации молекул воды близкая к 100 при обычных условиях при трехмерной организации ассоциатов из трехмерной превращается в линейную. Энергия взаимодействия между отдельными молекулами и кластерами невелика, но скорости, с которыми отдельные молекулы входят в кластеры или покидают их, достаточно высоки. Отсюда очень низкая вязкость флюида и чрезвычайно высокая диффузионная способность, а отсюда более высокая растворимость многих веществ и более высокая их подвижность. Так, использование аммиачного флюида позволяет получить кубическую модификацию BN из аморфной гексагональной при рекордно низких давлениях ~2 ГПа и 1500 К, в то время как «сухой» синтез с использованием специально приготовленных катализаторов осуществим только при давлениях не менее 4 ГПа. Синтез алмаза из графита в присутствии дигидрата щавелевой кислоты ((СОООН)2·2Н2О) размером до 30 мкм осуществляется в водно-двуокись углеродном флюиде, образующимся при разложении гидрата, при 7.7 ГПа и 1700 К без всяких катализаторов. Оба вещества не растворимы (без разрушения) ни в одном из известных растворителей. Вряд ли они целиком растворимы и в рассмотренных флюидах, но очень слабо, видимо, все же растворяются и этого оказывается достаточным для осуществления кристаллизации очень важных для современной техники сверхтвердых материалов.
При T>673 К все углеводороды, кислород, водород неограниченно смешиваются с водой, находящейся в сверхкритическом состоянии.
Основное внимание к этим средам проявляют технологи. Если еще совсем недавно щирокомасштабное использование флюидов даже не обсуждалось и было уделом лабораторий, то сейчас уже реализованы технологические процессы декофеинизации в углекислоте и деасфальтизации смазочных масел в субкритическом пропане путем экстракции ненужных или, наоборот, нужных веществ.
Процесс обратный экстракции – импрегнация в суперкритических растворителей используется для окрашивания полиэфирных волокон, например известного под торговой маркой «кримплен», и модифицирования поверхностей тормозных дисков для увеличения их износостойкости.
Совершенно уникальные возможности для исследователей представляет жидкостная сверхкритическая хроматография, позволяющая анализировать и очищать нелетучие и плохорастворимые вещества, например высшие фуллерены, выделение которых с применением обычных методов или крайне затруднено или просто невозможно.
Наиболее «старый» и хорошо известный метод синтеза совершенных монокристаллов ряда плохо растворимых, но широко использующихся в современной технике неорганических веществ, таких как SiO2, GeO2, AlPO4, Al2O3 и других, связан с т.н. гидротермальным синтезом, проводящимся в сверхкритической воде или воде близкой к этому состоянию. Собственно этот метод можно рассматривать как искусственно созданного дублера природного процесса, протекающего в недрах Земли на глубине примерно 50 км и ниже. Этот же метод используется в лабораториях для получения неорганических веществ – того же алмаза из силикатно-карбонатных растворов - и веществ в совершенно нетипичных для данного элемента валентных состояниях. Например, LnO2, LnCuO3, NaNiF6 и других. Осуществление этих реакций указывает на то, что вода в сверхкритических условиях существенно изменяет свои свойства. Действительно, из полярной жидкости она превращается в практически неполярную и при этом приобретает свойства сильного окислителя. Это приводит к тому, что в ней начинают растворяться многие органические вещества и при этом практически нацело окисляться. На этом свойстве в настоящее время разрабатываются технологии обезвреживания и ликвидации опасных для жизни отходов и веществ, таких как диоксины или искусственные ОВ. Однако из-за очень высокой окислительной способности флюидной воды, еще более возрастающей в присутствии галогенсодержащих веществ, настолько резко увеличивается ее коррозионная активность, что аппаратура, изготовленная из самых стойких конструкционных материалов, не выдерживает нескольких технологических циклов обезвреживания отходов.
Применение сверхкритических жидкостей для проведения химических реакций, хотя и используется, но в целом это все же экзотика, поскольку практическая реализация подобных технологий требует достаточно серьезного экономического обоснования. Известно несколько технологических процессов и реакций, в которых сверхкритический растворитель является одним из реагентов, например в гидрировании двуокиси углерода водородом с образованием муравьиной кислоты, в синтезе бутанола-2 путем присоединения воды к сверхкритическому бутену или в синтезе нитридов металлов из их гидридов или чистых металлов в сверхкритическом аммиаке.
Несколько иная организация конечной стадии процесса, связанная с заменой «штатного» растворителя на более летучий, с последующим быстрым стравливанием газовой фазы через форсунку (технология быстрого расширения сверхкритических растворов), позволяет получать пленочные материалы. Например, пленки селена, используемые для изготовления полупроводниковых фотодетекторов, очень высокого качества.
В последнее десятилетие довольно широкое распространение получили методы синтеза неорганических материалов на основе оксидов и реже сульфидов из специально приготовленных коллоидных растворов. Преимущества подобного подхода объясняются различными причинами, например возможностью совместить элементы, не сосуществующие в истинных растворах, например бор и кремний в виде боратов и силикатов, или в достаточно широких пределах изменять соотношения компонентов и их концентрацию, поскольку к коллоидным растворам в силу их неравновесности не применимо понятие «растворимость». Высушивая коллоидные растворы, нанесенные, например, на органические пленки, или в ходе их продавливания через фильеру, или просто в массе, можно получать самые различные материалы от ультрафильтров, и волокон до носителей для иммобилизации катализаторов с огромной поверхностью. Собирательное название этих методов – золь-гель технология, из которого видно, что процесс основан на переходе от коллоидного раствора (золя) к коллоидному осадку (гелю). Подобный прием без всяких кавычек можно отнести к области нанотехнологии.
Для получения золей обычно используют диспергационные и конденсационные методы. Во-первых, используют механические, ультразвуковые, электродинамические, т.е. преимущественно физические методы воздействия на вещество, приводящие к его измельчению, хотя при этом могут использоваться и химические методы синтеза и в особенности стабилизации золей. Например, при приготовлении золей трудно растворимых оксидов гидролиз солей проводят в присутствии оснований, способствующих пептизации (процесс обратный коагуляции) осадка гидроксида (гидроксополимерный метод) (уравнение 20).
Al(NO3)3 + 3 NH3 + 3 H2O ® {Al(OH)3 + 3 NH4NO3} (20)
Стабилизация золя (в устаревшей для этих веществ номенклатуре – мицеллы) осуществляется благодаря образованию в присутствии стабилизатора двойного электрического слоя, хотя известны примеры, когда этот слой самонаводится при взаимодействии поверхности дисперсной фазы с молекулами растворителя.
В конденсационных методах используют как физические эффекты, например конденсацию пара, замену растворителя или изменение растворимости, так и химические реакции, например реакцию гидролиза алкоголятов. Процесс «старения» продукта гидролиза приводит к получению незаряженных, но, конечно, окруженных молекулами растворителя и взаимодействующих с ними частиц полимерного (олигомерного) золя оксида металла. В этом случае определяющей в свойствах золя и затем геля является специфика химической реакции, в результате которой образуется полимер с различной длиной цепи, различной разветвленностью, способом сшивки с другими цепями и т.д., которые в конечном итоге определяют плотность сетки полимера. Для получения полимерного геля следует избегать присутствия в растворе электролитов, которые способствуют образованию и стабилизации мицелл. Именно поэтому наиболее часто используется реакция гидролиза алкоголятов металлов, в результате которой в качестве побочного продукта образуется только спирт, или растворы солей, пропущенные через ионнообменные смолы, на которых закрепляется анион кислотного осадка, что также эффективно предотвращает мицеллообразование. Для получения оксидного материала золь концентрируют тем или иным методом. Увеличение концентрации дисперсной фазы приводит к коагуляционным контактам между частицами и началу структурирования, т.е. образования геля. Его независимо от его природы подвергают операции «старения», которая для коллоидных растворов неизбежна в силу их неравновесности, затем сушат и, наконец, подвергают термообработке. Большая часть нужных свойства материала закладываются на стадиях синтеза и старения, хотя заметную роль играет и стадия термообработки, условия проведения которой дает возможность получать или рыхлый осадок с развитой поверхностью, используемый в гетерогенном катализе как носитель или самостоятельная фаза, или монолитный блок оксидной керамики, пригодной для изготовления лопаток турбин реактивных двигателей.
Твердофазные реакции. Через истинные растворы и золи мы дошли до твердого состояния. Твердофазные реакции считаются не только самыми «неэкономичными», но и достаточно «грубыми» с точки зрения возможности регулирования состава конечного продукта, исследования механизма превращения, получения чистого вещества и т.п. Тем не менее именно эти реакции несут пальму первенства в производстве большинства практически важных многотоннажных неорганических веществ и материалов и малотоннажных материалов высоких технологий.
«Грубость» твердофазных реакций вполне объяснима. В отличие от жидкофазных и тем более газофазных реакций, в которых взаимодействие протекает между строго определенным и ограниченным числом индивидуальных атомов, молекул, ионов или радикалов с ограниченным набором промежуточных продуктов, в ходе твердофазного взаимодействия в реакцию вступают агрегаты, содержащие большое число элементарных частиц, взаимодействующих друг с другом и влияющих на реакционную способность друг друга (кооперативные эффекты). Вследствие этого и число промежуточных веществ и стадий в этих реакциях, мало отличающихся друг от друга по энергиям, имеет или может иметь существенно больший набор.
Хорошо известно, что в газофазных и жидкофазных реакциях все пространство занимаемое реагентами – арена реакции. Одной из особенностей твердофазных реакций является их топохимический характер, т.е. локализация реакционной зоны на поверхности раздела фаз реагентов и продуктов реакции и отсюда резкая зависимость их скорости от размера реагирующих частиц..
Можно выделить три наиболее важных типа твердофазных реакций:
S1 ® S2 (21)
S1 + S2« S3 (22)
S1 + S2 ® S3 + S4 (23)
К превращениям первого типа относятся полиморфные превращения. Для большинства простых веществ это, в общем-то, скорее физические процессы, хотя для некоторых из них они могут протекать, как химические реакции.
Реакции второго типа довольно часто используют в химическом материаловедении для получения материала с функциональными свойствами, например в синтезе оксидной сверхпроводящей керамики. Реакции обратные синтезу также используются в материаловедении, например распад твердых растворов в системе Al-Fe-Ni-Co, полученных при высоких температурах, приводит к существенному увеличению коэрцетивной силы магнитов на основе этих сплавов. Другой пример реакций – распад комплексных тернарных нитридов щелочного, щелочноземельного металлов и бора по притектической реакции в ходе тремобарическго синтеза. Эта реакция приводит к кристаллизации кубического нитрида бора – аналога алмаза, но еще более устойчивого к высоким температурам и поэтому более интересного для машиностроителей в качестве режущего и абразивного материала (см. ниже).
Реакции третьего типа могут быть источником новых соединений. Например, обменные реакции гидридов щелочных металлов с галогенидами алюминия или бериллия в термобарическом синтезе в одну стадию приводят к получению термодинамически неустойчивых гидридов алюминия или бериллия, являющихся ценными компонентами топлив. Но могут быть и крайне нежелательными, например, для гетероструктур, которые в процессе эксплуатации могут разрушаться в результате появления новых фаз и терять все свои потребительские качества.
Активация твердофазных реакций обычно достигается за счет измельчения вещества (повышения площади возможных контактов), повышения температуры (увеличения скорости диффузии реагирующих веществ) и/или давления (экструзия, сдвиговые деформации и т.п.). Согласно Аррениусу влияние нагрева сводится к увеличению числа активных молекул, столкновение которых приводит к образованию нужного нам продукта. Доля активных столкновений a=hэф./h= e-E/RT, где hэф их число, h общее число столкновений, Е энергия активации, характеризующая величину энергетического барьера, обусловленную электростатическим отталкиванием электронных оболочек реагирующих частиц, который им надо преодолеть, чтобы произошло перераспределение связей. Так как скорость реакции пропорциональна числу эффективных столкновений в единицу времени, то k=k0-E/RT, где k – константа скорости реакции, k0 –предэкспоненциальный множитель. Отсюда следует, что скорость химической реакции будет возрастать с повышением температуры по экспоненте, а согласно эмпирическому правилу Ван-Гоффа при увеличении температуры реакции на 100 С скорость реакции возрастает в 2-4 раза (но это касается только области умеренных температур).
Высокие давления в трансформации вещества. Сверхтвердые материалы
Как отмечалось выше, при увеличении температуры скорость реакции возрастает по экспоненте. И если не рассматривать криогенные и туннельные реакции, протекающие около 77 К и вблизи 0 К, соответственно, то влияние температуры на направление и скорость химической реакции не требует особых пояснений. В то же время влияние концентраций реагирующих веществ и давлений, при которых осуществляется это взаимодействие, не столь однозначно для прогноза и понимания возможных путей превращения.
Давление в осуществлении химического взаимодействия может играть как положительную, так и отрицательную роль. Поскольку наибольший эффект при использовании высоких давлений наблюдается при осуществлении превращений в твердой фазе или системах «газ-твердое тело» и существенно меньшее в системах, содержащих жидкую(ие) фазу(ы) (в силу их меньшей сжимаемости), основное внимание мы будем уделять рассмотрению примеров из ряда гетерогенных систем. *)
Расчет термодинамических функций веществ и систем в различных агрегатных состояниях, находящихся под действием высокого давления, вопросы влияния давления на химическое равновесие и кинетику процессов достаточно подробно рассмотрены в
монографии Я.Я. Калашникова «Физическая химия веществ при высоких давлениях» М., Высшая школа, 1987. Здесь мы только кратко затронем общие вопросы влияния давления на химическое равновесие в конденсированных системах.
Как и в реальных газовых системах, где давления заменяется летучестями, т.е. вместо dG = RT d ln p используется dG = RT d ln f, так и в реальных жидких и твердых системах
*) Для характеристики величины давления используются различные единицы. В системе СИ принят паскаль, равный силе в 1 Н, приложенный к площади в 1 м2, т.е. 1Па=1Н/1 м2. Однако более распространены внесистемные единицы. Например, 1 бар=105Па и производные от него: 1 Кбар=108 Па = 100МПа; 1 Мбар=1022 Па=100ГПа. Соотношения между употребляемыми в литературе величинами следующие 105 Па=1 бар= 1.01972 кг/см2= 0.98692 атм= 750.06 мм рт.ст.; 1 атм=101325 Па=0.101325 МПа. В англоязычной особенно технической литературе применяется единица давления, выраженная в футах на квадратный дюйм (обозначение - psi)
(растворах) концентрации заменяются активностями и появляется выражение d μi = RT d ln ai, где μi химический потенциал i- компонента, ai –его активность. Константа равновесия для реакции mA + nB ↔ qC + rD выразится, как 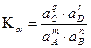 и будет зависеть только от температуры. При этом зависимость активности от давления определится уравнением
и будет зависеть только от температуры. При этом зависимость активности от давления определится уравнением 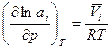 аналогичным уравнению, определяющему зависимость летучести от давления. Поскольку коэффициент активности
аналогичным уравнению, определяющему зависимость летучести от давления. Поскольку коэффициент активности 
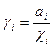 , (χi- молярная доля i-го компонента) то, подставляя его в уравнение константы равновесия, получаем
, (χi- молярная доля i-го компонента) то, подставляя его в уравнение константы равновесия, получаем 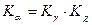 . Отсюда, так же, как и в случае химического равновесия в газовых
. Отсюда, так же, как и в случае химического равновесия в газовых
системах, получаем уравнение 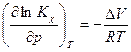 , где
, где 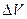 - изменение объема системы, происходящее при протекании реакции. Оно определяется экспериментально или
- изменение объема системы, происходящее при протекании реакции. Оно определяется экспериментально или
вычисляется по значениям парциальных молярных объемов реагентов. Однако, как правило, значения последних неизвестны, поэтому в рассмотрение вводится понятие идеального раствора и принимается, что парциальные молярные объемы веществ в системе равны молярным объемам чистых веществ. Тогда последнее уравнение запишется, как 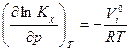 и активность i компонента под давлением можно вычислить по данным о сжимаемости чистого вещества i.
и активность i компонента под давлением можно вычислить по данным о сжимаемости чистого вещества i.
При высоких давлениях сжимаемость жидкостей и особенно твердых тел в зависимости от их природы может быть значительной. Поэтому пренебрежение этим параметром может привести к большим ошибкам в определении 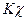 . Наиболее ярко этот эффект проявляется у веществ, активно реагирующих на величину приложенного давления (например, графита, превращающегося в более плотный алмаз). На практике эту реакцию характеризуют степенью сжатия
. Наиболее ярко этот эффект проявляется у веществ, активно реагирующих на величину приложенного давления (например, графита, превращающегося в более плотный алмаз). На практике эту реакцию характеризуют степенью сжатия 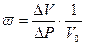 , где V0 – исходный объем при нормальных условиях (1 атм, 300К), DV – уменьшение объема в результате действия высокого давления в интервале давлений DP=Р-Р0.
, где V0 – исходный объем при нормальных условиях (1 атм, 300К), DV – уменьшение объема в результате действия высокого давления в интервале давлений DP=Р-Р0.
Также особенно велико влияние давления на равновесие, если процесс протекает с изменением количества вещества (например, при получении аммиака из водорода и азота, или гидрида алюминия из алюминия и водорода (правило Ле Шателье)). В этом случае прогноз о целесообразности использования техники высокого давления при проведении процесса делается из рассмотрения стехиометрии предполагаемой реакции.
Реакции типа 1 описывают полиморфные превращения. Для большинства веществ это, в общем-то, скорее физические процессы, обусловленные перестройкой геометрии молекулы и кристаллической структуры вещества. Для сложных и некоторых простых веществ полиморфные превращения могут сопровождаться разрывом старых и образованием новых связей, т.е. протекать как химические реакции. Различают энантиотропные - обратимые и монотропные - необратимыепревращения. Естественно, что монотропные модификации метастбильны, хотя многие из них кинетически могут существовать неопределенно долгое время (алмаз, черный фосфор). Это указывает на большое значение энергии активационного барьера, разделяющего метастабильную и стабильную фазы. Энантиотропные превращения протекают при вполне определенных и постоянных температурах и давлениях. Температура монотропных переходов не постоянна и зависит от предыстории образца и способа осуществления превращения.
Роль давления на характер протекания химических реакций, в том числе и фазовых переходов всех типов, изучена существенно хуже роли температуры или концентрации. Однако еще до развертывания широкомасштабных работ в этом направлении было вполне очевидно, что термобарическое воздействие на вещество может привести к таким радикальным изменениям в его фундаментальных свойствах, что в результате мы получим атом с принципиально иным типом гибридизации, иной реакционной способностью. Например, сжатие молекулярного азота на алмазных наковальнях до 150 GPa при одновременном лазерном нагреве образца до 2000 К приводит к образованию совершенно уникального вещества - полимерного азота с атомами, находящимися в sp3-гибридизации и связанными простыми σ-связями N-N. В кубической структуре этой фазы трехмерные цепи азота имеют гош-конформацию по отношению друг другу (рис. 40), но, будучи фазой высокого давления, полимерный азот при снятии нагрузки деполимеризуется и вновь становится обычным диазотом. Аналогичная обработка азида натрия приводит к образованию фазы с двухмерным полимерным анионом (N3)-n, также нестабильным при обычных условиях.
Также весьма интересно поведение дикислорода в условиях очень высоких давлений. При нормальном давлении молекулярный кислород затвердевает при температурах ниже 54,8 К. Это так называемая альфа-фаза твердого кислорода, в которой он существует до давлений порядка 0,1 ГПа. При комнатной же температуре кислород
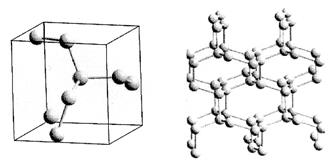
Рис. 40. Структура полимерного азота (слева – примитивная решетка, справа – размноженная, объемная).
затвердевает только при давлении порядка 4 ГПа и образует молекулярный кристалл в так называемой g-O2 фазе. При повышении давления твердый кислород испытывает ряд фазовых превращений: при давлении 5 ГПа он переходит в b-О2 фазу, при 7,5 ГПа - d -фазу, при 10 ГПа – в e-фазу или так называемый "красный кислород". Наконец, при давлении 96 ГПа, в x-фазе, твердый кислород становится металлом.
Структура «красного кислорода» установлена совсем недавно. Как видно из рис. 41, в узлах его кристаллической решетки располагаются молекулярные образования, состоящие из четырех молекул дикислорода. К сожалению, это образование также
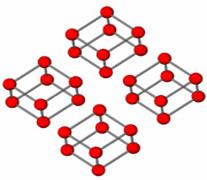
Рис. 41. Структура «красного кислорода» (e-фаза)
является фазой высокого давления и при снятии нагрузки распадается на привычный нам дикислород.
Весьма эффективно применение техники высоких давлений для осуществления фазовых переходов у веществ, имеющих многочисленные и легко деформируемые водородные связи. В этом отношении достаточно хорошо известен пример водяного льда. Как видно из рис. 42, при давлениях ниже 220 МПа и температуре ниже 0 оС вода существует в виде обычного природного льда I. Его объем больше объема жидкой воды и поэтому для него dT/dp<0. Это хорошо видно из рисунка, на котором кривая равновесия «лед I-вода» образует тупой угол с осью абсцисс и с ростом давления т.пл. льда понижается. При давлениях выше 220 МПа ΔV=Vж-Vт меняет знак, и лед I переходит в более плотную, чем жидкая вода модификацию. Для нее и вс
Дата добавления: 2016-07-18; просмотров: 4235;
Поиск по сайту
Узнать еще
- I. Реакции присоединения
- I. Реакции фенольного гидроксила
- I. Способы представления переменного синусоидального тока и напряжения.
- II. Реакции образования простых и сложных эфиров
- III. Методы искусственной физико-химической детоксикации.
- III. Реакции окисления
- III. Реакции по углеводородному радикалу
- IV. Влияние катализатора на скорость реакции

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
