КИСЛОТЫ И ОСНОВАНИЯ
В лекции по координационной химии мы в качестве курьеза привели попытку некоторых авторов «новых» подходов рассматривать химию, во всяком случае ее неорганическую часть, в терминах и с позиций координационной и показали, что эта попытка в подавляющем большинстве случаев, за исключением собственно химии комплексных соединений, не приводит к какому-либо существенному улучшению понятийного аппарата или новым открытиям. В то же время два может быть наиболее «древних» понятия в химии кислотаи основаниемогут претендовать на такое объединяющее начало, хотя и в очень широком смысле. Действительно, всю химию, все типы взаимодействия между веществами можно рассматривать, как реакции кислот с основаниями.
Уже с момента осознания существования двух противоположных по свойствам классов химических веществ (Р.Бойль) вплоть до новых времен происходит поиск всеобъемлющего определения этих понятий, поиск, расширяющий эти определения иногда вплоть до потери смысла, или, в ряде случаев, сужение областей их применимости (Г.Ф. Руэлль, А. Лавуазье, Ю. Либих, С. Аррениус). По Аррениусу-Оствальду кислотой является водородсодержащее соединение, которое в водном растворе диссоциирует с образованием ионов водорода, а основание, соответственно, гидроксилсодержащее соединение, которое в водном растворе образует гилроксил-ионы. Обязательным условием кислотно-основного взаимодействия по Аррениусу является протекание реакции нейтрализации по схеме 6
Н+ + ОН-« Н2О (6)
Сила кислоты и основания определяется способностью к диссоциации, т.е. константой диссоциации. Теория, неплохо объясняя большое число экспериментальных фактов, например причины различий в силе кислот, имеющих различные степени диссоциации, имеет и существенные ограничения. В нее, например, не вписываются аммиак и амины, являющиеся основаниями, но не имеющими гидроксильных групп, кислые и основные свойства соединений в неводных органических средах и растворителях донорного типа, амфотерность и многие другие экспериментально обнаруженные факты, которые не поддавались объяснению с позиций теории Аррениуса-Оствальда.
Первая попытка объединения теории водных и неводных растворов была сделана в рамках теории сольво-систем (сольвентная теория), развитой Э. Франклином, согласно которой кислоты и основания определяются как вещества, при растворении которых в растворе возрастает концентрация катионов и анионов по сравнению с чистым растворителем. Например, реакции автопротолиза воды и аммиака (уравнения 7,8) однотипны и образующиеся при их окончании ионы аммония и оксония выступают как кислоты.
Н2О + Н2О = Н3О+ + ОН- (7)
NH3 + NH3 = NH4+ + NH2- (8)
При растворении хлористого аммония в аммиаке концентрация ионов аммония возрастает против их концентрации в автопротолитической реакции, поэтому это вещество относят к сольво-кислотам. При растворении амида натрия возрастает концентрация анионов NH2-, поэтому это вещество относят к сольво-основаниям. Таким образом, сольво-кислоты и сольво-основания являются химическими соединениями, образующими катионы и анионы идентичные ионам растворителя, т.е. являются его производными. В этом случае реакция кислотно-основного взаимодействия выражается уравнением 9
Кислота + основание = соль + растворитель (9)
или (10)
(H3O)+Cl- + NaOH = NaCl + 2H2O (10)
Действительно, Э. Франклином и Х. Кади было показано, что растворы аммониевых солей в жидком аммиаке дают все реакции, характерные для водных растворов кислот (уравнения 11, 12):
2(H3O)+Cl- + Mg = MgCl2 + H2 + 2H2O (11)
2(NH4)+Cl- + Mg = MgCl2 + H2 + 2NH3 (12)
В общем случае в данной теории предполагается, что все кислоты и основания являются производными воды, аммиака, сероводорода и ряда других «типовых» веществ. Конечно, все эти представления сейчас, да и уже в момент появления казались странноватыми и фактически устаревшими, поскольку были опубликованы даже несколько позже более универсальной электронной теории Льюиса. Безусловно, положительным в ней является привлечение внимания к кислотно-основному взаимодействию в неводных средах. В то же время она оставалась строго ограниченной свойствами растворителя, причем растворителя с значительными константами аутоионизации, что является не таким уж типичным явлением.
Протонная теория Й. Бренстеда и Т.Лоури является дальнейшим развитием представлений о кислотно-основных равновесиях. Согласно ей – кислота является донором протона, а основание его акцептором. Ключевой вывод из этой теории: кислота не может существовать без основания, а основание без кислоты, участвующих в кислотно-основном взаимодействии, описываемом «двойным протолитическим уравнением» (13)
A1 + B2 « A2 + B1 (13)
или, например, (14)
СH3COOH + NH3 « NH4+ + CH3COO-. (14)
Для этой реакции NH4+ (А2) является сопряженной кислотой основания NH3 (В2), а CH3COO- (В1) сопряженным основанием кислоты СH3COOH (А1). Из этих уравнений очевидны предпосылки для оценки силы кислот и оснований: сильная кислота отдает протон легче, чем слабая, а сильное основание связывает его крепче, чем слабое. Равновесие (13) сдвигается вправо тем сильнее, чем сильнее кислота A1 и слабее кислота A2.
Двойное протолитическое уравнение можно представить в виде двух сопряженных равновесий, в которых кислоты и основания составляют сопряженные пары, связанные уравнениями 15 и 16 каждое с своими константами
НА « Н+ + А- (15)
В + Н+ « ВН+ (16)
Отсюда с очевидностью следует, что индивидуальное вещество не может быть само по себе кислотой или основанием. Ими они могут быть только по отношению друг к другу.
Собственно эти уравнения являются основой для определения силы кислот по шкале значений констант их диссоциаций в воде рКa, вычисляемых из закона действующих масс с учетом активности всех участвующих в равновесии частиц, т.е. с учетом «ионной атмосферы» и вообще всех типов взаимодействий «частица-растворитель». Величина отрицательного логарифма от pKa, т.е. [–lgKa], означает, что, чем меньше ее значение, тем более сильной является кислота. Если в обозначении используется просто Ka, то наоборот - чем больше значение константы, тем вещество проявляет более сильные кислотные свойства и, соответственно, меньше его основность.
Отрицательный логарифм от концентрации ионов водорода рН=-lg[H+] или, что более правильно, от их активности 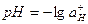 , иногда называемый водородным показателем, был введен в химическую практику в 1909 году С.П.Л. Зёренсеном. Нижней границе рН=0 соответствует разбавленный раствор серной кислоты с концентрацией 5 масс.%, вследствие чего измерить кислотность той же, но более концентрированной кислоты или вообще кислот более сильных, чем 5% серная кислота, данный метод оценки не позволяет.
, иногда называемый водородным показателем, был введен в химическую практику в 1909 году С.П.Л. Зёренсеном. Нижней границе рН=0 соответствует разбавленный раствор серной кислоты с концентрацией 5 масс.%, вследствие чего измерить кислотность той же, но более концентрированной кислоты или вообще кислот более сильных, чем 5% серная кислота, данный метод оценки не позволяет.
Вода в протонной теории выполняет двоякую функцию – функцию растворителя и сопряженного основания (уравнение 17)
HY +H2O «Y - + H3O+ (17)
Отсюда следует, что на положение равновесия будут оказывать сильнейшее влияние такие свойства растворителя, как его способность к сольватация ионов, и его диэлектрическая проницаемость (в последнем случае влияние e отмечается в случае различий в зарядах исходной и сопряженной кислот – при их равенстве e не оказывает заметной роли на величину константы). Однако при определении рКa помимо активности необходимо учитывать, что при растворении в воде многие кислоты диссоциируют нацело, т.е. все они превращаются в ониевые соли - аддукты кислоты и растворителя, например H3O+Cl- или NH4+Cl- (последний, как известно, является устойчивым соединением и может быть выделен в индивидуальном состоянии). Это уже вторая причина (первая – концентрационная, см. выше), почему в подобных растворителях невозможно оценить силу кислот относительно друг друга. Иными словами, вода оказывает на них нивелирующее действие и все кислоты более сильные, чем Н3О+, в ней равны по силе, а в аммиаке даже слабые органические кислоты диссоциированы нацело, т.е. в нем почти все кислоты образуют ониевые соли. Аналогично и основния в достаточно кислых растворителях обладают одинаковой основностью. Поэтому для оценки кислотных или основных свойств таких веществ необходимо применять менее основный или, соответственно, менее кислый т.н. дифференцирующий растворитель. Например, в метаноле азотная кислота диссоциирована частично, а хлористоводородная нацело. В ацетоне или фтористом водороде все кислоты за исключением хлорной – слабые электролиты. В чистом виде уксусная кислота, например, является гораздо более слабой кислотой, чем ее раствор в воде.
Здесь нет противоречия с предыдущим, когда отмечалось, что само по себе вещество не может быть ни кислотой, ни основанием, поскольку большинство растворителей сами обладают кислотными или основными свойствам, например за счет вышеотмеченной реакции автопротолиза. Так, уксусная кислота или вода сами же акцептируют свои протоны (уравнение 17):
СH3COOH + СH3COOH « СH3COO - + СH3COOH2+ (17)
Такие растворители или вещества, в отличие от апротонных неспособных к самодиссоциации и, таким образом, не проявляющих или проявляющих очень слабые кислотные или основные функции, называются амфипротонными. Следует особо отметить, что понятия апротонные и инертные растворители отнюдь не идентичны и их применение в качестве синонимов грубейшая ошибка, поскольку парафины, например, в определенных условиях могут быть донорами протонов, т.е. кислотами, о чем мы поговорим в разделе C-H кислоты, а арены их акцепторами, т.е. быть основаниями.
Традиционные кислоты в зависимости от свойств партнера могут менять свою функцию и вести себя как доноры или акцепторы протона, например по уравнению 18
СH3COOH + HNO3 « NO3- + CH3COOH2+ (18)
В пространстве теории Бренстеда-Лоури вещества, присоединяющие протоны, называются протофильными и являются основаниями (это относится и к веществам, являющимися растворителями, например, аммиак и амины, причем, чем к большему числу веществ они относятся как основания, тем они сильнее как основания). Соединения, отдающие протоны, называются протогенными и являются кислотами. Если они к тому же выполняют функцию растворителя, то их называют протогенными растворителями. Для них, как и для оснований, такая же зависимость – чем легче они отдают протоны, т.е. чем они сильнее как кислоты, тем большее количество веществ в этих растворителях проявляют основные свойства.
Следует иметь ввиду, что все рассмотренные процессы являются схематическими. В действительности механизм кислотно-основного взаимодействия в бренстэдовском смысле более сложен и включает стадию образования комплексов с водородными связями типа АН…В. В ряде случаев оно на этом и заканчивается. При более благоприятных условиях, например в присутствии растворителя с высокой диэлектрической проницаемостью, происходит полное протонирование основания и завершение реакции. Это указывает и на дополнительное взаимодействие компонентов с растворителем, т.е. их сольватацию, которая существенно влияет на относительную силу кислоты. Важен и другой аспект, связанный с изменением свойств лигандов координированных к металлическому центру. Например, вода в комплексе [CrPy2(H2O)2(OH)2]Cl проявляет ярко выраженные кислые свойства. Аналогичные явление наблюдается для молекулы аммиака в комплексе [Pt(NH3)4(NH3)Cl]Cl2, что объясняется связыванием неподеленных пар электронов у гетероатомов и потерей ими основных свойств.
Более общей является электронная теория кислот и оснований, предложенная Льюисом. В этом случае к кислотам относятся вещества, способные использовать неподеленную пару электронов другого атома для создания устойчивой электронной группировки одного из своих атомов (т.е. фактически любые вещества, имеющие вакантные орбитали), а к основаниям – вещества, обладающие неподеленной парой электронов, которая может быть использована для образования такой электронной группировки. Наиболее часто такой группировкой оказывается октет электронов. Взаимодействие этих веществ также протекает как кислотно-основное взаимодействие, но уже в льюисовском смысле и приводит к образованию донорно-акцепторной или какой-либо другой более слабой электростатической связи и соответственно комплексных соединений различной природы.
Кислоты и основания в электронной теории классифицируют по типу орбиталей, принимающих участие в образовании связи. Все кислоты или акцепторы разделяются на s, n и p-типы, а основания или доноры на n, s и p-типы. В образовании связи принимает участие наиболее высокая занятая граничная молекулярная орбиталь (ВЗМО) основания и наиболее низкая по энергии вакантная орбиталь кислоты (НВМО). В соответствии с типом орбиталей классифицируют 9 типов комплексов: nn (R3N.MeXn), ns (R3N.I2), sn (RX.MeXn), ss (RX.I2), sp (RX.ArH), pn (ArH.MeXn), ps (ArH.I2) и pp (ArH.тетрацианохинодиметан), (Ме- металл). При этом следует иметь ввиду, что, как и в протонной теории, в зависимости от условий и свойств партнера одно и то же соединение может выступать и в качестве основания и как кислота.
Понятие основание в теориях Брэнстеда и Льюиса совпадает, понятие кислоты в теории Льюиса более широкое и протонные кислоты в ней рассматриваются как протоны, нейтрализованные основаниями. В объединяющей оба подхода еще более общей теории М.Усановича к кислотам относят вещества, способные отдавать катионы или присоединять анионы, а к основаниям вещества способные отщеплять анионы или присоединять катионы. В этом случае уже практически все химические реакции, включая окислительно-восстановительные, но с определенными ограничениями, можно рассматривать с позиций кислотно-основного взаимодействия, а продукты их взаимодействия как комплексы или аддукты льюисовских кислот и оснований. Именно из этого положения Р. Пирсоном были введены понятия жестких, мягких и промежуточных кислот и соответствующий им принцип ЖМКО.
При оценке «мягкости» и «жесткости» кислот и оснований учитывают их химический состав, электронное строение и устойчивость образованных ими в результате кислотно-основного взаимодействия комплексов.
Мягкие основания – доноры, обладающие высокой поляризуемостью, низкой электроотрицательностью, легко окисляются, их ВЗМО имеют высокую энергию (H-, I-, CN-, CO, R3P(As,S),RS- и т.д.).
Жесткие основания – доноры с низкой поляризуемостью, высокой электроотрицательностью, трудно окисляются, их ВЗМО имеют низкую энергию (H2O, OH-, ROH, RO-, R2O, NH3, F-, Cl-, SO4-2 и т.д.).
Мягкие кислоты – акцепторы с высокой поляризуемостью, низкой электроотрицательностью и небольшим зарядом, легко восстанавливаются и имеют высоколежащие НВМО (Cu+, Ag+, Hg+, BH3, R+, I2, Br2, I+, ICN и т.д.)
Жесткие кислоты – акцепторы с низкой поляризуемостью, высокой электроотрицательностью и зарядом, трудно восстанавливаются, незаполненные НВМО имеют низкую энергию (H+, Li+, Mg+2, Al+3, Ln+3, AlH3, HНal и т.д.)
Взаимодействие между этими веществами протекают таким образом, что ЖК преимущественно реагирует с ЖО, а МК с МО. Этот экспериментальный факт объясняется тем, что взаимодействие между орбиталями с близкой энергией более эффективно, чем с орбиталями существенно различающимся по энергии.
Таким образом, более чем полуторовековой поиск классификационных признаков привел к достаточно полному определению «кислота – основание». Но не менее важен другой аспект этих поисков, а именно понимание того, что какими бы словами мы не называли наблюдаемое химическое явление, или вещество, или частицу, принимающих участие в химической реакции: кислота-основание, донор-акцептор, нуклеофил-электрофил, электрон-дырка, анион-катион, т.е. терминами взятыми из самых разных областей химии и физики, – все это объединяется общим понятием «кислотно-основное взаимодействие», а на бытовом уровне принципом «вилка-розетка» или еще более бытовом «папа-мама».
В заключение этой темы следует остановиться на рассмотрении некоторых необычных для «стандартной» химии понятий, которые, однако, достаточно широко используются на уровне специалистов. Эти понятия связаны с RН-кислотами и суперкислотами
RН-кислоты. В соответствии с определением Бренстэда все кислоты его типа должны иметь в своем составе протоны. Органические вещества за редким исключением отвечают этому требованию. Другое дело будет ли органическое соединение обладать заметными кислотными свойствами. А это в основном определяется двумя факторами – различиями в электроотрицательностях между атомом водорода и связанной с ним атомом или группой атомов, например H-CH3 и H-Cl, размерами последних, определяющими степень их поляризуемости или способности к делокализации электрона, например H-F < H-I или H-OH < H-SeH и внешним фактором, например основностью сореагента. Органические вещества, в которых под действием подходящего основания или какого-либо подходящего физического воздействия, например фотолиза, может происходит гетеролиз связей С-Н, О-Н, S-H, N-H и Si-H, приводящий к образованию протона и карбаниона, в органической химии называют CH, NH, OH и SH - кислотами. В этом же ряду возрастает и их сила. В химии для их активации как кислот обычно пользуются подходящим основанием, растворенным в хорошем, т.е. сильноосновном растворителе. Их выбор осуществляют по величинам рК СН-кислоты в этом растворителе и соединения, образующегося при присоединении протона в этом же растворителе. Например, натриевая соль ацетилена может быть получена в жидком аммиаке по реакции 19
NH3 жид.
HC≡CH + Na+NH2- ® HC≡C- Na+ + NH3 (19)
pK 25 pK 33
Эту реакцию можно рассматривать как реакцию кислоты (ацетилена) с солью еще более слабой кислоты –амидом натрия.
В органической химии известно понятие «протонный гарпун», получившее название из-за способности некоторых сильных оснований ионизировать слабые кислоты по связям С-Н. Например, алкиламидлития R2NLi в реакции с 4,6-дихлорпиримидином-Cl, имеющем подвижные атомы хлора, вступает не в реакцию их нуклеофильного замещения на амидную группу, а в реакцию металлирирования по связи С-Н, приводящей к образованию металлорганического литиевого производного.
С-Cl
N C-H
H-C C-Cl
N
Суперкислоты. Выше мы говорили, что сила кислот Брэнстеда определяется по константе их диссоциации в воде, которая выполняет функцию растворителя и сопряженного основания. Очевидно, что в случае кислот полностью диссоциирующих в этом растворителе невозможно оценить их силу. Поскольку основность растворителя определяет степень диссоциации, для оценки силы таких кислот необходимо подобрать менее основный растворитель. В общем случае для сравнения силы кислот в фиксированной системе растворитель-основание (B+S) используется функция кислотности Гаммета Но. Согласно постулату Гаммета «Существует серия слабых оснований - индикаторов, отношение активности ионизированной и неионизированной форм которых не зависит от природы индикатора, а определяется только кислотностью среды». К таким основаниям относится один из представителей ряда нитробензола, варьирование заместителей в котором позволяет изменять основность в пределах 20 порядков, не меняя типа образующихся в результате реакции протонирования анилиниевых катионов.
Принято считать, что область суперкислот простирается от 100% для серной кислоты с Но=-12 и ниже, например фторсульфоновая HSO3F кислота с Но=-15.1. Еще более сильные кислоты образуются при взаимодействии протонных кислот, имеющих атомы с неподеленными парами электронов, т.е. могущих выступать как основания Льюиса, с некоторыми сильными кислотами Льюиса, например олеум – соединение серной кислоты с серным ангидридом или соединение фторсульфоновой кислоты с пентафторидом сурьмы, романтически названной «магической кислотой». В последнем случае мы имеем комплексное соединение состава H[SO3{FSbF5}], обладающее настолько мощными кислыми свойствами, что растворяет парафины, т.е. последние в ней выступают как сопряженные основания. Еще большей кислотностью обладают смеси фтористого водорода с пентафторидом сурьмы. Так, если во всех других известных суперкислотах при добавлении воды сразу же происходит образование катиона оксония, который не претерпевает реакций обмена с дейтерозамещеной водой, то в кислоте {HF+SbF5}, реакция дейтерообмена легко осуществляется. Этот факт объясняют появлением у иона оксония, хотя и слабых, но основных свойств, приводящим к его протонировнию и образованию дикатиона Н4О+2! Более того, в суперкислых растворах протон может присоединяться к метану, образуя катион метония CH5+. Безусловным достижением химиков является синтез веществ, содержащих катионы Xe2+ и N5+ с противоионами SbF6-, Sb2F11-и AsF6-, соответственно. Естественно, что парафины также будут более подвержены атаке этими веществами и их крекинг, протекающий с разрывом связей С-С и С-Н, может быть осуществлен уже при комнатной температуре. Хорошо известная в органическом синтезе реакция Фриделя-Крафтса – реакция алкилирования ароматических углеводородов в присутствии смесей галогенидов алюминия, галоидводородов или галоидалкилов, также обусловлена образованием в ходе реакции суперкислот и их последующим каталитическим действием. Во многих промышленно важных каталитических процессах, протекающих в рамках кислотно-основного катализа (крекинг нефтей, их гидрирование, алкилирование и т.п.) используются кислые гетерогенные катализаторы минерального ряда (цеолиты, оксиды), или полимерные материалы (Nafion-H –полностью фторированный полиэтилен). Все они готовятся на базе сформированных представлений о суперкислотных центрах, т.е. модифицированных в зависимости от их исходной функции обработкой или кислотами Льюиса или сильными минеральными кислотами. Пример близкий к работам нашей кафедры – интеркаляты фторидов сурьмы или мышьяка, хлоридов алюминия и железа в межслоевое пространство графита. Все эти соединения в той или иной мере проявляют свойства суперкислот, хотя их практическое применение в отличие от чисто минеральных фаз затруднено из-за протекания сложных химических реакций, приводящих к постепенному разрушению углеродной матрицы, ковалентному связыванию продуктов реакции и, как следствие, к дезактивации каталитических свойств.
С чем же связно такое резкое возрастание кислых свойств этих веществ? Объяснение очень простое - протон фактически не удерживается объемным комплексным анионом даже в отсутствие сольватирующей среды. То, что это действительно так, нам демонстрирует синтез одно- и двухосновных «карборановых кислот» состава Н(СНВ11Сl11) и H2(B12X12) (X=Cl, Br) с икосаэдрическим углеродным каркасом, чрезвычайно устойчивым к действию многих химических реагентов. Именно поэтому, несмотря на очень сильные кислотные свойства (предполагается, что на сегодняшний день это самые сильные кислоты из известных суперкислот способные протонировать
бензол и даже некоторые инертные газы), «карборановые кислоты» довольно инертны своей анионной частью, которая не вступает или вступает, но с трудом в дальнейшие превращения.
Дает ли нам право вышеизложенное предположить, что аналогичную ситуацию можно организовать и с основаниями, т.е. получить супероснование? Наверное, можно, но для этого следует использовать объемный некоординирующий или слабо координирующий катион.
Дата добавления: 2016-07-18; просмотров: 2440;
Поиск по сайту
Узнать еще
- А - при выпучине; б - при просадке основания; 1 - выпучина; 2 - отверстие; 3 -гидрофобный грунт; 4 - наладка; 5 - просадка основания; 6 - сварной шов; 7 -днище.
- А. ОДНООСНОВНЫЕ ПРЕДЕЛЬНЫЕ КИСЛОТЫ
- АМИДЫ СУЛЬФАНИЛОВОЙ КИСЛОТЫ (СУЛЬФАНИЛАМИДНЫЕ ПРЕПАРАТЫ)
- Б. ОДНООСНОВНЫЕ НЕПРЕДЕЛЬНЫЕ КИСЛОТЫ
- Биополимеры: белки, нуклеиновые кислоты.
- В норме аминокислоты, всосавшиеся в кровь из кишечника, циркулируют в крови 5 — 10 мин и очень быстро поглощаются печенью и частично другими органами (почками, сердцем, мышцами).
- В. ДВУХОСНОВНЫЕ КИСЛОТЫ ЖИРНОГО РЯДА
- Виды приговоров и основания их постановления

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
