КООРДИНАЦИОННАЯ ХИМИЯ
По определению, данному Вернером, к комплексным (координационным) соединениям относятся молекулярные соединения, не показывающие в растворах всех свойств исходных для них соединений первого порядка. Последние сейчас называны симплексами. При этом «атомы в КС могут проявлять не только главную, но дополнительную или побочную валентность; в комплексном ионе имеется центральный атом (не обязательно ион металла, это может быть нейтральный атом или анион), вокруг которого располагаются координированные им ионы и/или молекулы, называемые лигандами или аддендами; каждый комплекс характеризуется координационным числом и такими понятиям, как внутренняя и внешняя сферы и центральное ядро. В первоначальном своем смысле к предмету химии координационных соединений относили молекулярные соединения, обладающие высокой прочностью. Соединения менее прочные, сильно диссоциирующие в растворах, относили к двойным солям. Молекулярные комплексы, пи-комплексы, КПЗ из-за отсутствия их в каталогах науки начала XX века естественно не рассматривались. В настоящее время определение понятия КС сильно изменилось и к ним относят практически все вещества, составленные из простых веществ, даже несмотря на то, что по своим свойствам и строению они не подходят под правила Вернера, определяющие понятие и условия образования комплексов. К таким соединениям относится, например, большое число молекулярных комплексов, сильно диссоциированных в растворе, большая часть КПЗ и другие молекулярные комплексы. Более того, появилась тенденция рассматривать даже такие вещества как обычные соли, типа поваренной, но, правда, только в кристаллическом состоянии, с точки зрения или в терминах координационной химии (Я.А. Угай). Согласно принципа Оккама такой подход - это изобретение лишней сущности, хотя в ряде случаев он оправдан и уж во всяком случае, не криминален, поскольку не нарушает правил образования КС, сформулированных Вернером.
Координационная связь, образованная между лигандом и металлом-комплексообразователем, может быть различной по своей природе – ионная, полярная любого происхождения, донорно-акцепторная, ковалентная, дативная или какая-либо другая, а также одинарная, двойная или тройная, а в биядерных или полиядерных комплексах переходных металлов возможна кратная связь между атомами металлов. Число связей, образованных одним и тем же лигандом или число связей, которое он может образовать, с одним атомом металла-комплексообразователя, определяет дентатность лиганда. Лиганды могут быть моно-, ди- или би-, а в общем случае полидентатными. Если в лиганде имеется два или более донорных центра, причем все они координируются к одному атому, то это хелатирующие или хелатные лиганды, а образующиеся при этом комплексы - хелаты, если полидентатный лиганд присоединен к разным центрам, то это мостиковый лиганд. В случае, если в одном лиганде есть два разных по природе донорных центра, например атомы азота и серы, то лиганд может дополнительно называться амбидентатным. Согласно Вернеру связь между лигандом и атомом-комплексообразователем, образованная только за счет донирования электронов с лиганда, называется дополнительной или побочной в противоположность главной, которая изначально присутствует в центральной составляющей комплекса. Сейчас мы знаем, что это далеко не так и подобное разделение может фиксироваться только в очень небольшом числе молекулярных комплексов с гетеролептическими лигандами, например, в недавно описанном катионе [NH3Cl]+, стабилизированным фторсодержащими анионами, типа BF4 или SbF6. В подавляющем же большинстве случаев после комплексообразования уже нельзя различить какая связь главная, а какая побочная, поскольку, как уже отмечалось выше, в случае присоединения гомолигандов все связи усредняются и имеют равные геометрические и энергетические характеристики, в случае гетеролигандов связи различны, но выделить среди них главную и побочную можно только ретроспективно.
В настоящее время используются три подхода к описанию химической связи в комплексах. В порядке усложнения и глубины выводов это – метод валентных связей, теория кристаллического поля и теория поля лигандов.
Исторически первым был метод валентных связей, предложенный Полингом. В этом методе образование комплекса представляется как реакция между кислотой Льюиса (центральный атом) и основанием Льюиса (лиганды) с формированием за счет гибридизации атомных орбиталей (отсюда и вышеотмеченное равенство связей) только ковалентных (донорно-акцепторных) s-связей. При этом возможна реализация трех основных типов гибридизации и соответственно образование трех типов координационных многогранников: октаэдра - sp3d2 , тетраэдра - sp3 и квадрата - dsp2. Так, атом Pt в ст. окисления +2 с d8 электронной конфигурацией, т.е. атом с 8 электронами на внешней валентной оболочке[¯][¯][¯][¯][ ] [ ] [ ][ ][ ], в комплексном анионе [PtCl4]2- имеет плоско-квадратное окружение, поскольку комплекс диамагнитен и заполнение электронных уровней в нем следующее: ¯¯¯¯¯(5d) ¯(6s) ¯¯(6p) (dsp2 - конфигурация). [Cr(NH3)6]+3 должен быть и есть парамагнитен, поскольку в d3 -электронной конфигурации невозбужденного иона Cr+3 имеется три неспаренных электрона, располагающихся на трех 3d подуровнях и не меняющих своего положения при комплексообразовании (спин-свободный или высокоспиновый комплекс), и частично вакантные и полностью вакантные 4s и 4р – орбитали, которые в ходе реакции комплексообразования занимаются шестью парами электронов или ¯ ¯(3d) ¯(4s) ¯¯¯(4p) (d2sp3-гибридизация, октаэдр). В невозбужденном ионе Co+3, находящегося в электронной конфигурации d6, пять d-орбиталей заняты шестью электронами, 4 из которых неспарены, а 4s и 4p –орбитали вакантны. Поскольку комплекс [Co(NH3)6]+3 диамагнитен (спин-спаренный или низкоспиновый комплекс), необходимо, чтобы четыре неспаренных электрона спарились, освободили две d-ячейки, что приведет к реализации октаэдрического окружения с d2sp3-гибридизацией ¯¯¯¯¯(3d) ¯(4s) ¯¯¯(4p). Однако не все так гладко, поскольку сразу же были найдены вещества, не вписывающиеся в эту простую схему. Это, например, парамагнитные высокоспиновые (или спин-свободные) комплексы [CoIIIF6]3- (d6 конфигурация) или [Ni IICl4]2- (d8 конфигурация). Для объяснения их электронного строения были предложены следующие схемы заполнения электронных уровней: ¯ (3d) ¯(4s) ¯¯¯(4p) ¯ ¯(4d), sp3d2- гибридизация, октаэдр, и ¯¯ ¯ (3d) ¯(4s) ¯¯¯(4p), sp3 - гибридизация, тетраэдр, соответственно, т.е. схемы с незаполненными нижележащими и энергетически более выгодных 3d уровнями. Именно это свойство приводит к отличию строения комплекса никеля от строения плоского квадрата, наблюдаемого в изоэлектронном хлор-комплексном анионе платины+2. По мнению многих ученых это достаточно плохо обоснованная гипотеза. Но в ряде случаев она достаточна для качественного описания строения комплекса и его магнитных свойств и поэтому явилась причиной разделения комплексов на внутриорбитальные, в которых электроны лигандов размещаются на внутренних вакантных орбиталях, и внешнеорбитальные, в которых электроны лигандов переходят на более высокие по энергии уровни.
Лучшие результаты и соответственно большее распространение получила теория поля лигандов и ее простейший вариант - теория кристаллического поля.
В ТКП взаимодействие между центральным атомов и лигандами в отличие от МВС рассматривается как чисто электростатическое, без привлечения понятий гибридизации орбиталей, донорно-акцепторных связей и без учета размера и электронного строения лигандов. Иными словами принимаются в учет только распределение в пространстве d или f- орбиталей центрального атома и их электростатическое отталкивание друг от друга, а лиганды рассматриваются как точечные отрицательные заряды или диполи, размещенные в пространстве таким образом, чтобы энергия их взаимного отталкивания достигала минимальных значений. При этом постулируется и другое отличие от метода валентных связей – в комплексе орбитали центрального атома отталкиваются, а не притягиваются, как в МВС, от точечных зарядов лиганда и поэтому стремятся расположиться в пространстве таким образом, чтобы их взаимодействие было минимальным. Этот постулат предполагает только электростатическое взаимодействие электронов лигандов и центрального атома, ковалентные силы не учитываются.
В силу свойств симметрии для четырехлепестковой формы d-орбиталей возможны не 6, а только пять волновых функций - dz2 (является линейной комбинацией dz2-y2 и dz2-x2 орбиталей), dyz, dxz, dxy и dx2-y2 c граничными поверхностями орбиталей, показанными на рисунке 2. Все они энергетически равноценны, т.е. являются вырожденными. При подходе лигандов к центральному атому они, в соответствии с принципом минимального электростатического взаимодействия, располагаются либо в вершинах октаэдра, либо тетраэдра и реже квадрата, а электроны центрального атома в первую очередь занимают те орбитали, которые максимально удалены от участков сферы, где сосредоточен заряд лигандов. Таким образом, возникает неэквивалентность d-орбиталей и вырождение снимается. В случае октаэдрических комплексов принимают, что направление орбиталей dz2 и dx2-y2 совпадает с направлением осей октаэдра, в вершинах которого располагаются лиганды. Эти энергетически невыгодные дважды вырожденные орбитали обозначаются символом eg. Трижды вырожденные орбитали dyz, dxz, dxy принимают положение между осями и мало взаимодействуют с электронами лиганда, т.е. являются более энергетически выгодными и обозначаются символом t2g. Прямо противоположная картина предполагается в случае тетраэдрических комплексов – с направлением на лиганды совпадают оси орбиталей dyz, dxz и dxy, обозначаемые символом t2g, а удалены и энергетически боле выгодными являются две орбитали: dz2 и dx2-y2, обозначаемые символом eg. В результате подобного перераспределения электронной плотности появляются различия в энергиях орбиталей центрального атома и общее вырождение снимается (рисунок). Разность энергии между eg и t2g , называемой энергией расщепления d-подуровня, обозначается как 10Dq или D. Для каждого электрона в октаэдрических комплексах энергия eg-орбиталей возрастает до 6Dq или 3/5D против исходно вырожденного, а энергия t2g-орбиталей уменьшается до 4Dq или 2/5D. Для тетраэдрических комплексов все наоборот при этом во всех случаях суммарно энергетический центр тяжести d-орбиталей не изменяется.
Проблема спаренных и неспаренных электронов в данной теории также решается с энергетических позиций. Если энергия необходимая для спаривания (Р) меньше, чем D, образуются низкоспиновые комплексы, больше – высокоспиновые. Соотношение этих величин и соответственно образование того или иного типа комплексов, в первую очередь определяется природой и зарядом центрального атома и природой лиганда. Так, ионы с полузаполненной d5-конфигурацией (Fe+3, Mn+2) имеют очень высокое значение Р и поэтому они как правило высокоспиновые, а ионы с d6-конфигурацией имеют вдвое меньшие энергии спаривания, поэтому для них типичны низкоспиновые комплексы. Величина энергии расщепления D больше для высокозарядных катионов (за счет увеличения электростатического отталкивания орбиталей от лигандов), чем для низкозарядных и, в целом, возрастает от периода к периоду от 3d к 5d-металлов. Далее, она меньше в тетраэдрических комплексах, чем в октаэдрических, поэтому первые из-за меньшей вероятности того, что P будет <D, чаще бывают высокоспиновыми. Наконец, определяющим в какой ячейке окажется комплекс является природа лиганда, которые располагаются в спектрохимический ряд I-<Br-<Cl-<F-<C2O4-2 £H2O<NCS<py~NH3<en<dipy<NO2-<CN-»CO. По величине влияния на энергию расщепления лиганды делятся на лиганды слабого поля (обычно левее аммиака, дают высокоспиновые комплексы, поскольку Р<D) и сильного поля (низкоспиновые комплексы).
В результате комплексообразования достигается тот или иной выигрыш энергии, который называют энергий стабилизации кристаллическим полем (ЭСКП). Эта энергия максимальна, когда электроны центрального атома размещаются на наиболее низких подуровнях данного уровня, подвергшегося расщеплению под влиянием поля лигандов. Так, в поле октаэдрической симметрии максимальный выигрыш в энергии достигается при заполнении t2g-подуровня, а подуровень eg остается вакантным (сильное поле конфигурация t2g6). Если заполнены оба подуровня, то ЭКСП=0 (слабое или сильное поле, конфигурация t2g6 eg4), тоже будет наблюдаться и для октаэдрических высокоспиновых комплексов (слабое поле) с конфигурацией атома t2g3 eg2 и т.д.
В случае веществ, в которых возможно равновероятное заполнение t2g или eg подуровней, например в комплексах Cu(II) или Ni(II) с конфигурацией t2g6 eg3 и в которых электроны на eg подуровнях могут располагаться в комбинациях 1+2 или 2+1, происходит тетрагональное искажение правильной октаэдрической геометрии, в пределе превращающее ее в квадрат. Это свойство объясняется эффектом Яна-Теллера, доказавшими теорему: «вырожденное электронное состояние всякой молекулярной нелинейной системы является неустойчивым, поэтому система подвергается искажению, понижающему ее симметрию и снимающему вырождение». Наиболее сильно этот эффект проявляется в комплексах с электронными конфигурациями t2g6 eg3 , t2g6 eg2 , t2g eg3 и t2g6 eg1, т.е. преимущественно в комплексах с вырождением на eg-орбиталях.
Объяснив причину и величину электронных спектров поглощения, теория кристаллического поля дала инструмент предсказательной силы для прогноза и объяснения строения классических координационных соединений.
Еще более совершенна в этом отношении, но и более сложна для поверхностного описания теория поля лигандов, которая использует методы молекулярных орбиталей в ТКП. Поэтому мы отметим только некоторые особенности и принципы на которых строится эта теория.
1. Образование комплексов и снятие вырождения происходит не только за счет электростатического взаимодействия, как постулируется в ТКП, но и за счет перекрывания орбиталей центрального атома и лигандов, т.е. и в рамках ковалентного взаимодействия (например лиганд сильного поля СО не имеет заряда и заметного дипольного момента, но образует прочные комплексы).
2. Взаимодействие двух атомных орбиталей дает две новых молекулярные орбитали: связывающую, лежащую ниже меньшей по энергии АО, и разрыхляющую, лежащую выше высшей по энергии АО, одновременно может наблюдаться и несвязывающая МО.
3. Взаимодействуют только атомные орбитали одинаковые по симметрии. Если таковых нет, то АО остаются несвязывающими.
4. Обязательное соблюдение принципа Паули и правил Хунда.
Применение данной теории позволяет расширить ее применение на p-комплексы, но рассмотрение этого расширения уже не входит в наши задачи.
В заключение этого раздела рассмотрим принцип блочного строительства координационных и, как их частный случай, кластерных соединений на основе принципа изолобальности, разработанного Р. Хоффманом.
При синтезе, например Cr(CO)6, из атома хрома, имеющего шесть валентных электронов на девяти атомных орбиталях (одна 4s, три 4p и пять 3d), и шести молекул СО, каждая из которых поставляет пару валентных электронов на одной орбитали, образуется шесть связывающих, три несвязывающих и шесть разрыхляющих МО. На первых двух располагаются 18 электронов, а разрыхляющие орбитали остаются вакантными (рис.) Процессы окисления, восстановления и другие превращения молекул прежде всего затрагивают высшие занятые молекулярные орбитали (ВЗМО) и нижние вакантные молекулярные орбитали (НВМО), которые называют граничными. Если энергия ВЗМО молекулы достаточно велика и находящиеся на ней электроны легко взаимодействуют с вакантными НВМО другой молекулы, то первые из них относятся к основаниям Льюиса (ОЛ), а вторые к кислотам Льюиса (КЛ). При наличии на одной из ВЗМО одного электрона возникает радикал R*, способный быть и ОЛ и КЛ, а при отсутствии двух электронов реализуется молекула, которая также в зависимости от партнера может быть и кислотой и основанием, в том числе и внутримолекулярными. Их комбинация приводит к образованию новых, координационных соединений. Грубо говоря, в данном подходе каждая из реагирующих молекул, как бы они ни были просты или сложны, рассматриваются как «черные ящики», в одном из которых есть неподеленная пара(ы) электронов или неспаренный электрон, а в другом вакантная орбиталь(и) (принцип «папа-мама») и которые подходят друг другу по «размеру». Отсюда возникло понятие «изолобальность» (от англ. lobe – доля, половинка орбитальной восьмерки). Два фрагмента считаются изолобальными (обозначение ), если их самые высокие по энергии орбитали имеют одинаковую симметрию (например σ-симметрия 1s орбитали атома водорода и sp30 для фрагментов, граничные орбитали которых сходны по числу, энергии, симметрии, направленности в пространстве и по заполненности электронами. При этом геометрия фрагментов их состав не рассматриваются («черный ящик» или блок, заготовка, встраиваемые в нужное место).
Один из простейших вариантов – одна из ВЗМО вакантна. Наиболее известна комбинация ОЛ®КЛ, например AlCl3NR3. В отсутствие молекулы ОЛ ее роль может выполнять электронная пара заместителя у центрального атома металла, например BCl3. При этом процесс «нейтрализации» происходит внутримолекулярно за счет переноса электронной плотности с атомов хлора на атом бора, что приводит к уплощению молекулы и частично кратным связям B-Cl, или межмолекулярно, например, в случае AlCl3, что приводит к образованию димера. Более того, даже, если заместители не имеют неподеленной пары электронов, например, в молекулах AlR3 или BH3, электронодефицитный центральный атом «забирает» часть электронной плотности и за счет ее делокализации реализуется двухэлектронное трехцентровое связывание.
В качестве наиболее распространенной электронодефицитной частицы выступает протон, имеющий вакантную сферическую s-орбиталь. На ней легко размещается пара электронов от одной или даже двух молекул воды. Аналогично протонируются и атомы переходных металлов в комплексах и кластерах, при этом в зависимости от соотношения электроотрицательностей могут образоваться ионные соединения или гидриды.
Особый интерес представляет сходство свойств протона и фрагмента Ph3PAu+. У атома золота для образования химической связи обычно используется только один электрон, расположенный на sp-орбитали. В катионе Ph3PAu+ она остается вакантной, что сообщает ему свойства очень похожие на свойства протона, в том числе и свойство «протонирования» КЛ. Это свойство позволило в последние годы весьма продвинуть МОХ золота.
Другой не очень сложный вариант – одна из ВЗМО полузаполнена. В этом случае изолобальными фрагментами являются такие радикальные частицы, как ·Н, ·СН3, ·NH2, ·OH,·F (последние три с одно, двумя и тремя неподеленными парами электронов). Их комбинации приводят к получению самых разнообразных молекул с этаноподобными ординарными связями, например H2N-NH2, НО-ОН, RS-SR. Геометрия радикалов, содержащих переходные металлы, радикально отличается от геометрии радикалов, построенных с участием непереходных элементов. Это объясняется участием в связывании d-электронов и соответствующих орбиталей. Тем не менее, если мы сможем сконструировать радикал с одной полузаполненной ВЗМО, то он будет изолобален радикалам, состоящим из непереходных элементов, например ·Mn(CO)5 (производное от Cr(CO)6 - без одной СО группы, но на один электрон больше у металла), который, в свою очередь, изолобален ·Со(СО)4. Отсюда он, как и ОН-радикал может димеризоваться, давая (CO)5Mn-Mn(CO)5, H2N-Mn(CO)5 или F-Mn(CO)5. Эти комбинации могут быть самыми разнообразными и приводить к образованию самых экзотичных соединений, но, конечно, существуют и ограничения, например стерические: как нельзя заменить в метане все четыре атома водорода на амино-группы, так и, например, в тетрахлорсилане нельзя заменить все атомы хлора на ·Со(СО)4 – группы.
Аналогичные закономерности наблюдаются и в случае фрагментов, имеющих две полузаполненные ВЗМО. К ним относятся карбены, нитрены с одной неподеленной парой электронов или атом кислорода с двумя такими парами. Их объединение приводит к появлению молекул с этиленоподобными связями типа собственно этилена, диимина RN=NR, формальдегида Н2С=О, нитрозосоединения RN=O и т.п. В соответствии с моделью Найхольма-Гиллеспи-Киперта прочность этих и вышерассмотренных соединений, точнее двойных связей, уменьшается с увеличением в составе молекул атомов с неподеленными парами электронов, а также при переходе к элементам 3 периода, имеющих диффузные валентные орбитали. Так, молекула кислорода вообще существует в виде бирадикала с одинарной связью О-О. Среди металлсодержащих фрагментов изолобальным бирадикалам является фрагмент Fe(CO)4. Однако комплексы с его участием, как правило, нестабильны и регистрируются в матрице инертного газа.
Изолобальные фрагменты с тремя полузаполненными ВЗМО образуют соединения с ацителеноподобными связями. Однако все биядерные частицы из этого ряда за исключением ацетилена и азота, как правило, малостабильны, но стабилизируются или при образовании тетрамеров (Р4, Со4(СО)12) или за счет координации с более объемными фрагментами, например Cp(CO)2WºCR.
Фрагменты с тремя вакантными граничными орбиталями, например V(CO)3-, Cr(CO)3, Mn(CO)3+ могут насыщаться за счет координации молекул, являющихся донорами шести электронов, например, триаминов или псевдоароматических и ароматических углеводородов типа С6Н6, С5Н5-, С3Н33-, дающих структуры типа «фортепианной табуретки».
Ввиду большого разнообразия типов координационных соединений не имеет смысла в рассмотрении подробной классификации этих веществ. Можно лишь отметить наиболее известные классы комплексов. Прежде всего это хелатные комплексные соединения, к которым также относятся внутрикомплексные соединения и комплексонаты металлов, аммино- и аминокомплексы, ацидокомплексы, ацидоаминокомплексы. В каждом из этих классов группируются десятки и сотни подвидов комплексных соединений, хотя в действительности их существенно больше, посколькув списке типичных нет, например, аутокомплексов, комплексов с сера- и фосфорсодержащими лигандами, алкильными, арильными, гидридными и p-лигандами, молекулярных комплексов и многих других.
Реакции комплексообразования приводят к заметному или даже существенному изменению химических свойств составляющих их компонентов. Таким образом, например, осуществляют стабилизацию необычных валентных состояний металлов, прежде всего металлов в низших ст. окисления вплоть до 0, наблюдаемых во многих карбонилах или металлорганических соединениях, и даже –1 для щелочных металлов в алкалидах (вещь немыслимая еще в начале XX века, поскольку противоречила всем имевшимся представлениям в теории химической связи), или, напротив, в высших ст.окисления вплоть до +6 для комплексных фторидов железа или +4 для РЗМ помимо Се. Естественно, что при этом меняются и химические свойства лигандов. В ряде случаев происходит стабилизация молекул лиганда, меняется их структура, усиливаются кислотные свойства и повышается реакционная способность по отношению к нуклеофильным веществам. Так, показана стабилизация молекулы альдимина Ph(OH)CH=NH во внутренней сфере комплексов, в свободном состоянии распадающегося на молекулу аммиака и салициловый альдегид. Отмечается заметное увеличение степени гидролиза координированных эфиров аминокислот, нитрилов, галогеналкиламинов и ряда других лигандов. Происходят реакции декарбоксилирования поликарбоновых кислот, кардинальная перестройка структуры лиганда, например фосфористой кислоты Н3РО3, в которой атом фосфора не имеет неподеленной пары электронов, в фосфин :P(OH)3, обладающий свойствами n-донора, и т.д.
Изменение электронной ситуации в комплексном соединении делает ряд из них каталитически активными в самых разнообразных реакциях. Именно на этом свойстве основан и в настоящее время интенсивно развивается металлокомплексный катализ и прежде всего гомогенный катализ. Например, координация этилена к платине с первоначальным образованием p-комплекса в присутствии воды приводит к каталитическому окислению олефина и образованию ацетальдегида. На этом принципе основан промышленный процесс синтеза этого важного для современной химической технологии вещества. Другой пример – координация галогенидом ПМ триаминных или оксоиминных лигандов приводит к комплексам с 14-ти электронным окружением атома металла. При их активации метилалюмоксаном происходит метиллирование атома металла и образование ионного комплекса [L,L’ М(CH3)+][L”-] с формально 12-ти электронным окружением атома металла в катионе, проявляющего высокую каталитическую активность в реакции полимеризации олефинов совершенно несвойственную для исходных галогенидов.
С самим лигандом также происходят изменения. При этом возможно как увеличение его реакционной способности, так и ее уменьшение, т.н. маскирование функциональной группы. Этот прием в органической химии используется для защиты реакционной групп от побочной или в настоящий момент нежелательной реакции. Таким образом, например реакцией Cu(II) с диаминкарбоновой кислотой (уравнение 2) была замаскирована a-аминогруппа и осуществлена реакция комплекса с мочевиной. После удаления меди сероводородом в индивидуальном состоянии был выделен труднодоступный цитруллин.
O (CH2)3NH2 O (CH2)3NH-C-NH2
С С С С O
O NH2 + NH2-C-NH2 ® O NH2 + NH3 (2)
Cu O Cu
И, наконец, совершенно замечательная реакция характерная только в химии комплексных соединений – реакция, называемая реакцией темплатного или темплейтного синтеза, о который мы уже немного говорили выше, и связанная с самосборкой сложной органической молекулы из более простых на матрице, в качестве которой обычной выступает ион металла. Классическим и наиболее простым примером такого синтеза является получение фталоцианина из динитрила фталевой кислоты С6H4(CN)2 в присутствии меди+2 (уравнение 3).
Обычно все же темплатные синтезы протекают более сложным путем, через стадию образования заготовок из исходного сырья для последующей их конденсации. Реакция
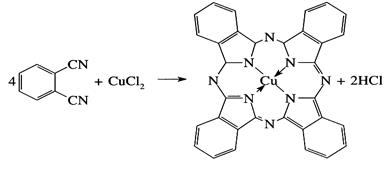 (3)
(3)
синтеза заготовок также происходит с участием иона металла, например салициловый альдегид в аммиачной среде и в присутствии никеля сперва претерпевает конденсацию Шиффа, приводящую к образованию металлодихелата, который уже и вступает в реакцию с аммиаком, давая циклический аминоэфир (уравнение 4).
Видно, что темплатный синтез приводит к получению макроциклических молекул, т.е. молекул содержащих в структуре не менее 4 гетероатомов. Количество лигсонов (так называют органическую молекулу, играющую роль одного из строительных
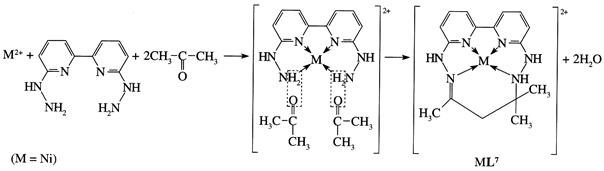
(4)
блоков макроцикла) в темплатном синтезе может варьировать от одного до 4-5, например, при синтезе дикатенана (уравнение 5):
Наиболее эффективны для проведения этой реакции ионы никеля+2, меди+2 и реже железа+2, которые имеют наиболее подходящие размеры для вхождения в полость образующегося хеланта (так называют конечный продукт темплатного синтеза), подходящие координационные числа для координации гетероатомов макроциклической молекулы и подходящую электронную структуру.
Несколько иной подход к объяснению предпочтительности этих атомов заключается в привлечении теории жестких и мягких кислот по Пирсону: для рассматриваемых ионов, относящихся к жестким и умеренно жестким кислотам, типична плоская ромбическая или тетраэдрическая координация лигандов, а в подавляющем большинстве макроциклических
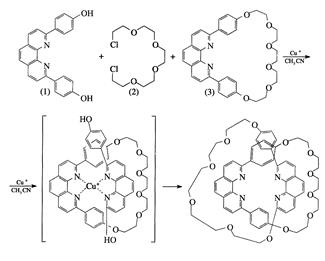
(5)
лигандов, получаемых в рамках темплатного синтеза, характерно присутствие именно четырех донорных центров, располагающихся в одной плоскости. Развитие метода темплатного синтеза позволяет достаточно просто получать очень сложные соединения, играющие важную роль в приготовлении люминесцентных материалов, фотоприемников, фотосенсибилизаторов, радиотерапевтических препаратов для диагностики раковых заболеваний и т.д.
Дата добавления: 2016-07-18; просмотров: 2785;
Поиск по сайту
Узнать еще
- Аналитическая химия и химический анализ. Предмет и задачи аналитической химии. Классификация методов химического анализа.
- БИОЛОГИЧЕСКАЯ ХИМИЯ
- Биоорганическая химия
- БИОХИМИЯ ЗУБНОГО НАЛЕТА. ЗУБНОЙ НАЛЕТ И КАРИЕС.
- Биохимия осложнений сахарного диабет.
- БИОХИМИЯ СОЕДИНИТЕЛЬНОЙ ТКАНИ
- ГЕОХИМИЯ КАК НАУКА. ПРЕДМЕТ И МЕТОД ГЕОХИМИИ
- Естественноисторический подход и эволюционная химия

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
