Биоорганическая химия
Биоорганическая химия – наука, изучающая строение и свойства веществ, участвующих в процессах жизнедеятельности, в непосредственной связи с познанием их биологических функций.
Биоорганическая химия наука изучающая строение и реакционную способность биологически значимых соединений. Предметом биоорганической химии являются биополимеры и биорегуляторы и их структурные элементы.
К биополимерам относятся белки, полисахариды (углеводы) и нуклеиновые кислоты. В эту группу также включают липиды, которые не являются ВМС, но в организме обычно связаны с другими биополимерами.
Биорегуляторы – это соединения, которые химически регулируют обмен веществ. К ним относятся витамины, гормоны, многие синтетические соединения, в том числе лекарственные вещества.
Биоорганическая химия базируется на идеях и методах органической химии.
Без знания общих закономерностей органической химии, сложно изучение биоорганической химии. Биоорганическая химия тесно связана с биологией, биологической химией, медицинской физикой.
Совокупность реакций, протекающих в условиях организма, называется метаболизмом.
Вещества, образующиеся в процессе метаболизма, называются – метаболитами.
Метаболизм имеет два направления:
Катаболизм – реакции распада сложных молекул на более простые.
Анаболизм - это процесс синтеза сложных молекул из более простых веществ с затратой энергии.
Термин биосинтез применяется по отношению к химической реакции IN VIVO (в организме), IN VITRO (вне организма)
Существуют антиметаболиты - конкуренты метаболитов в биохимических реакциях.
Сопряжение, как фактор повышения стабильности молекул. Взаимное влияние атомов в молекулах органических соединений и способы ее передачи
План лекции:
Сопряжение и его виды:
p, p - сопряжение,
r,p - сопряжение.
Энергия сопряжения.
Сопряженные системы с открытой цепью.
Витамин А, каротины.
Сопряжение в радикалах и ионах.
Сопряженные системы с замкнутой цепью. Ароматичность, критерии ароматичности, гетероциклические ароматические соединения.
Ковалентная связь: неполярная и полярная.
Индуктивный и мезомерный эффекты. ЭА и ЭД – заместители.
Основным типом химических связей в органической химии являются ковалентные связи. В органических молекулах атомы соединены s и p - связями.
Атомы в молекулах органических соединениях соединены ковалентными связями, которые называются s и p - связями.
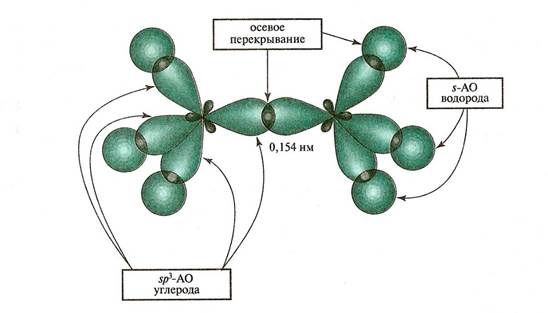
Одинарная s - связь в SP3 – гибридизованном состоянии характеризуется l – длиной (С-С 0,154 нм) Е-энергией ( 83 ккал/моль), полярностью и поляризуемостью. Например:
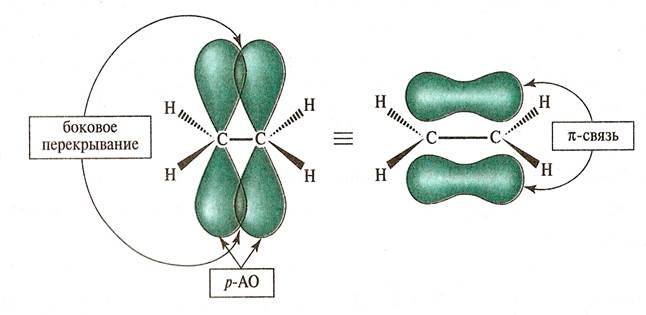
Двойная связь характерна ненасыщенным соединениям, в которых, кроме центровой s - связи, есть еще перекрывание перпендикулярное s - связи которая называется π-связью).
Двойные связи бывают локализованными, то есть электронная плотность охватывает только 2 ядра связываемых атомов.

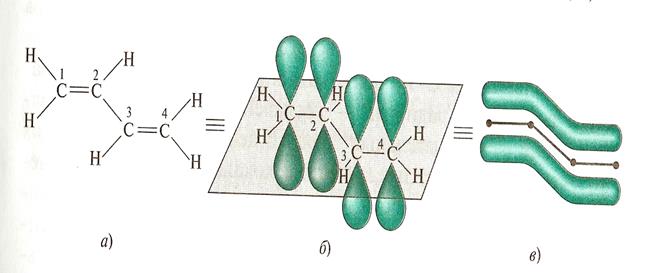
Чаще всего мы с вами будем иметь дело с сопряженными системами. Если же двойные связи чередуются с одинарными связями (а в общем случае у атома соединенного с двойной связью, есть р-орбиталь, то р-орбитали соседних атомов могут перекрываться друг с другом, образуя общую p - электронную систему). Такие системы называются сопряженными или делокализованными. Например: бутадиен-1,3
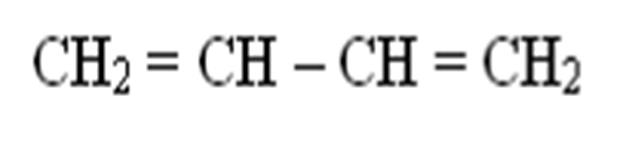
p, p - сопряженные системы
Все атомы в бутадиене находятся в SP2 – гибридизированном состоянии и лежат в одной плоскости (Рz – не гибрид орбитали). Рz – орбитали параллельны друг другу. Это создает условия их взаимного перекрывания. Перекрывание Рz орбитали происходит между С-1 и С-2 и С-3 и С-4, а также между С-2 и С-3, то есть возникает делокализованная ковалентная связь. Это находит отражение в изменении длин связей в молекуле. Длина связи между С-1 и С-2 увеличена, а между С-2 и С-3 укорочена, по сравнению с одинарной связью.
l-C -С, 154 нм l С=С 0,134 нм
l С-N 1,147 нм l С =O 0,121 нм
r, p - сопряжение
Примером р, π сопряженной системы может служить пептидная связь .
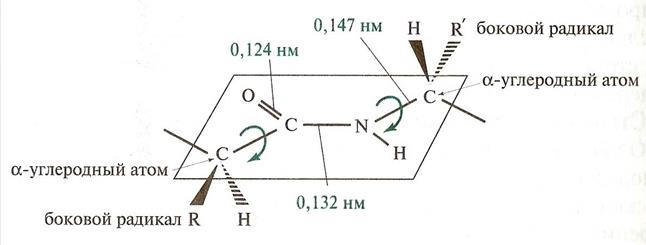
r, p - сопряженные системы
Двойная связь С=0 удлиняется до 0,124 нм против обычной длины 0,121, а связь С – N становится короче и становится равной 0,132 нм по сравнению с 0,147 нм в обычном случае. То есть процесс делокализации электронов приводит к выравниванию длин связей и снижению внутренней энергии молекулы. Однако ρ,p – сопряжение возникает в ациклических соединениях, не только когда чередуется = связи с одинарными С- С связями ,а еще при чередовании с гетероатомом:
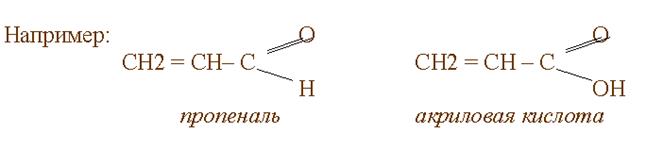
Рядом с двойной связью может находиться атом Х, имеющий свободную р- орбиталь. Чаще всего это гетероатомы О,N, S и их р-орбитали, взаимодействуют с p - связями, образуя р, p - сопряжение.
Например:
СН2 = СН – О – СН = СН2
Сопряжение может осуществляться не только в нейтральных молекулах, но и в радикалах и ионах:
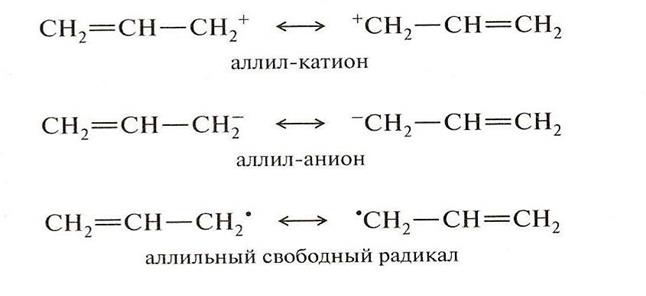
Исходя из выше изложенного, в открытых системах сопряжение возникает при следующих условиях:
Все атомы, участвующие в сопряженной системе, находятся в SP2 – гибридизованном состоянии.
Рz – орбитали всех атомов перпендикулярны плоскости s - скелета, то есть параллельны друг другу.
При образовании сопряженной многоцентровой системы происходит выравнивание длин связей. Здесь нет «чистых» одинарных и двойных связей.
Делокализация p-электронов в сопряженной системе сопровождается выделением энергии. Система переходит на более низкий энергетический уровень, становится более устойчивой, более стабильной. Так, образование сопряженной системы в случае бутадиена – 1,3 приводит к выбросу энергии в количестве 15 кДж/моль. Именно за счет сопряжения повышается устойчивость радикалов ионов аллильного типа и их распространенность в природе.
Чем длиннее цепь сопряжения, тем больше выброс энергии ее образования.
Это явление довольно широко распространено в биологически важных соединениях. Например:
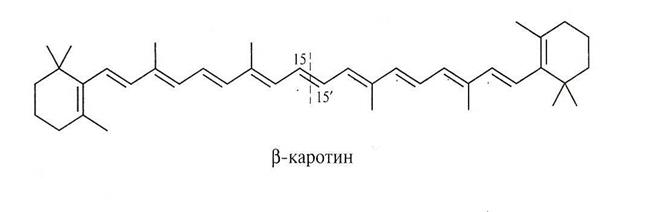
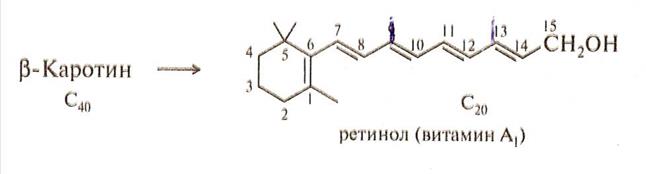
С вопросами термодинамической устойчивости молекул, ионов, радикалов мы будем постоянно встречаться в курсе биоорганической химии, к которым относятся ряд ионов и молекул широко распространенных в природе. Например: 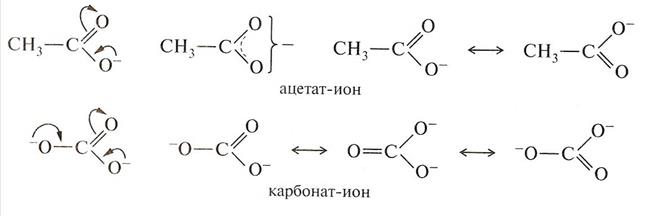
Сопряженные системы с замкнутой цепью
Ароматичность. В циклических молекулах при определенных условиях может возникать сопряженная система. Примером p, p - сопряженной системы является бензол, где p - электронное облако охватывает атомы углерода, такая система называется – ароматической.
Выигрыш энергии за счет сопряжения в бензоле составляет 150,6 кДж/моль. Поэтому бензол устойчив термически до температуры 900оС.
Наличие замкнутого электронного кольца доказано с помощью ЯМР. Если молекулу бензола поместить во внешнее магнитное поле, возникает индуктивный кольцевой ток.
Таким образом, критерием ароматичности, сформулированным Хюккелем является:
молекула имеет циклическое строение;
все атомы находятся в SP2 – гибридизованном состоянии;
существует делокализиванная p - электронная система, содержащая 4n + 2 электронов, где n – число циклов.
Например: 
Особое место в биоорганической химии занимает вопрос ароматичности гетероциклических соединений.
В циклических молекулах, содержащих гетероатомы (азот, сера, кислород) единое p - электронное облако, образуется с участием р – орбиталей атомов углерода и гетероатома.
Пятичленные гетероциклические соединения
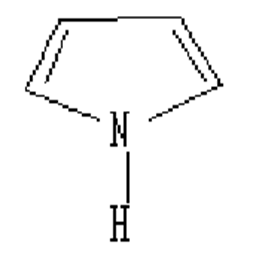
Ароматическая система образуется при взаимодействии 4-х р-орбиталей С и одной орбитали гетероатома, на котором находится 2 электрона. Шесть p - электронов образуют ароматический скелет. Такая сопряженная система является электронно избыточной. В пирроле атом N находится в SP2 гибридизированном состоянии .
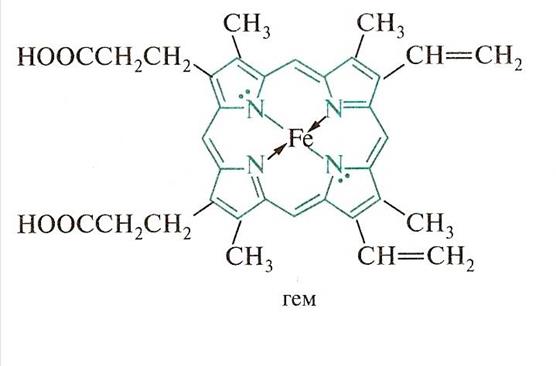
Пиррол входит в состав многих биологически важных веществ. Четыре пиррольных кольца образуют порфин – ароматическую систему с 26 p - электронами и высокой энергией сопряжения (840 кДж/моль)
Порфиновая структура входит в состав гемоглобина и хлорофилла 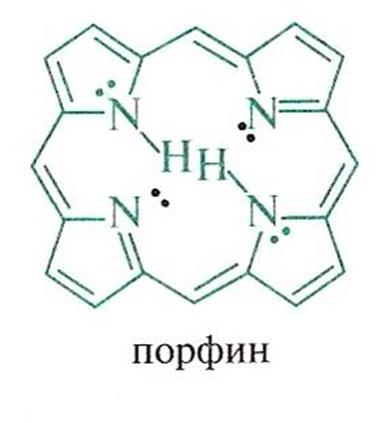
Шестичленные гетероциклические соединения
Ароматическая система в молекулах этих соединений образуется при взаимодействии пяти р-орбиталей атомов углерода и одной р-орбитали атома азота. Два электрона на двух SP2 – орбитали участвует в образовании s - связей с атомами углерода кольца. Р-орбиталь с одним электроном входит в ароматический скелет. SP2 – орбиталь с неподеленной парой электронов лежит в плоскости s - скелета.
Электронная плотность в пиримидине смещена к N, то есть система обеднена p - электронами, она электронно дефицитна.
Многие гетероциклические соединения могут содержать один и более гетероатомов 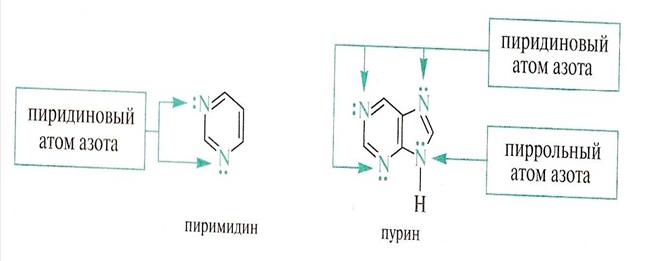
Ядра пиррола, пиримидина, пурина входят в состав многих биологически активных молекул.
Взаимное влияние атомов в молекулах органических соединений и способы его передачи
Как уже отмечалось, связи в молекулах органических соединений осуществляются за счет s и p связей, электронная плотность равномерно распределена между связанными атомами только тогда, когда эти атомы одинаковы или близки по электроотрицательности. Такие связи называются неполярными. 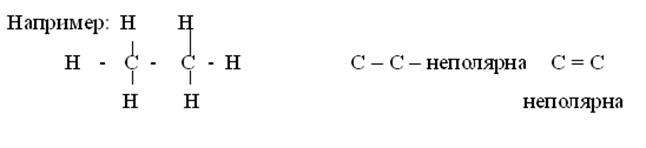
CH3-CH2→CI полярная связь
Чаще в органической химии имеем дело с полярными связями.
Если электронная плотность смешена в сторону более электроотрицательного атома, то такая связь называется – полярной. Основываясь на значениях энергии связей, американский химик Л.Полинг предложил количественную характеристику электроотрицательности атомов . Ниже представлена шкала Полинга.
Na Li H S C J Br Cl N O F
0,9 1,0 2,1 2,52,5 2,5 2,8 3,0 3,0 3,5 4,0
Атомы углерода в разном состоянии гибридизации различаются по электроотрицательности. Поэтому s - связь между SP3 и SP2 гибридизованными атомами - полярна 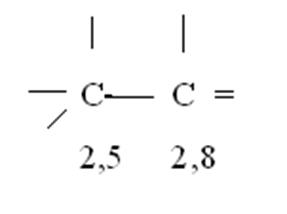
Индуктивный эффект
Передача электронной плотности по механизму электростатической индукции по цепи s - связей называется индукцией, эффект называется индуктивным и обозначается J. Действие J, как правило, затухает через три связи, однако близко расположенные атомы испытывают довольно сильное влияние находящегося рядом диполя.
Заместители, смещающие электронную плотность по цепи s - связей в свою сторону, проявляют -J – эффект, а наоборот +J эффект. 
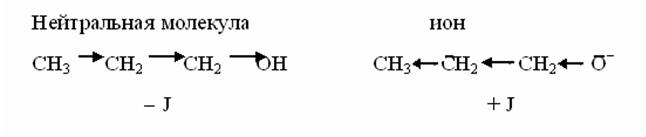
Изолированная p - связь, а также единое p - электронное облако открытой или замкнутой сопряженной системы способна легко поляризоваться под влиянием ЭА и ЭД заместителей. В этих случаях индуктивный эффект передается на p - связь, поэтому обозначает Jp. 
Мезомерный эффект (эффект сопряжения)
Перераспределение электронной плотности в сопряженной системе под влиянием заместителя, являющегося участником этой сопряженной системы, называется мезомерным эффектом (М-эффект). 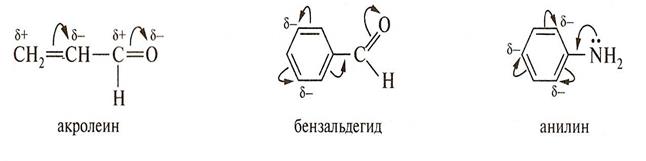
Для того, чтобы заместитель сам входил в сопряженную систему, он должен иметь либо двойную связь (p,p -сопряжение), либо гетероатомом с неподеленной парой электронов (r,p - сопряжение). М – эффект передается по сопряженной системе без затухания.
Заместители, понижающие электронную плотность в сопряженной системе (смещенная электронная плотность в свою сторону) проявляют -М-эффект,а заместители, повышающие электронную плотность в сопряженной системе проявляют+М-эффект.
Электронные эффекты заместителей 
Реакционная способность органических веществ в значительной степени зависит от характера действия J и M эффектов. Знание теоретических возможностей действия электронных эффектов позволяет предсказать ход тех или иных химических процессов.
Кислотно-основные свойства органических соединений Классификация органических реакций.
План лекции
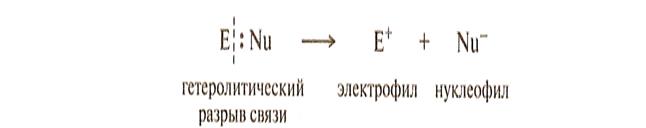
Понятие субстрата, нуклеофила, электрофила.
Классификация органических реакций.
обратимые и необратимые
радикальные, электрофильные, нуклеофильные, синхронные.
моно- и бимолекулярные
реакции замещения
реакции присоединения
реакции элиминирования
окисление и восстановление
кислотно-основные взаимодействия
Реакции региоселективные, хемоселективные, стереоселективные.
Реакции электрофильного присоединения. Правило Морковникова, антиморковниковское присоединение.
Реакции электрофильного замещения: ориентанты 1-го и 2-го рода.
Кислотно-основные свойства органических соединений.
кислотность и основность по Бренстеду
кислотность и основность по Льюису
Теория жестких и мягких кислит и оснований.
Классификация органических реакций
Систематизация органических реакций позволяет свести многообразие этих реакций к сравнительно не большому числу типов. Органические реакции можно классифицировать:
по направлению: обратимые и необратимые
по характеру изменения связей в субстрате и реагенте.
Субстрат – молекула, которая предоставляет атом углерода для образования новой связи
Реагент - действующее на субстрат соединение.
Реакции по характеру изменения связей в субстрате и реагенте можно разделить на:
радикальные R
электрофильные Е
нуклеофильные N (Y)
синхронные или согласованные 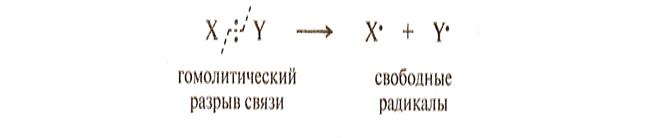
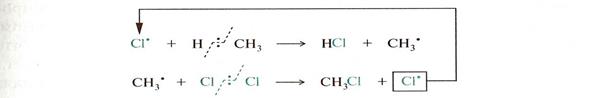
Механизм реакций SR
Инициирование
Рост цепи
Обрыв цепи 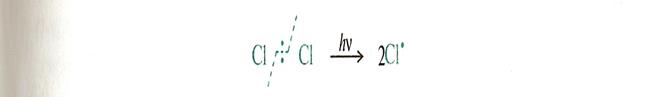
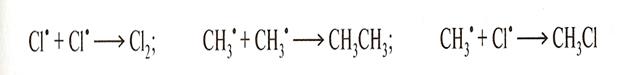
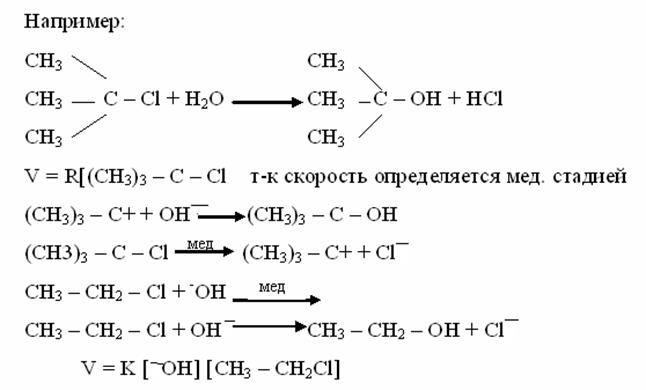
КЛАССИФИКАЦИЯ ПО КОНЕЧНОМУ РЕЗУЛЬТАТУ
Соответствие с конечным результатом реакции бывают:
А) реакции замещения
SR SE SN
Б) реакции присоединения
AR AE AN
В) реакции элиминирования
E
Г) перегруппировки
Д) окисление и восстановление
Е) кислотно-основные взаимодействия
Реакции также бывают:
Региоселективные – предпочтительно протекающие по одному из нескольких реакционных центров.
Хемоселективные – предпочтительное протекание реакции по одной из родственных функциональных групп.
Стереоселективные – предпочтительное образование одного из нескольких стереоизомеров.
Реакционная способность алкенов, алканов, алкадиенов , аренов и гетероциклических соединений
Основу органических соединений составляют углеводороды. Мы будем рассматривать лишь те реакции, осуществляемых в биологических условиях и соответственно не с самими углеводородами, а с участием углеводородных радикалов.
К ненасыщенным углеводородам мы относим алкены, алкадиены, алкины, циклоалкены и ароматические углеводороды. Объединяющее начало для них π – электронное облако. В динамических условиях также органические соединения склонны подвергаться атаке Е+
Однако, реакции взаимодействия для алкинов и аренов с реагенами приводит к разным результатам ,так как , в этих соединениях разная природа π – электронного облака: локализованная и делокализиванная.
Рассмотрение механизмов реакций начнем с реакций АЕ. Как нам известно, алкены взаимодействуют с 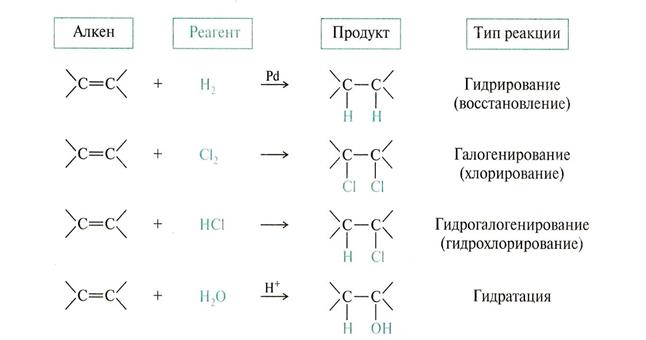
Механизм реакции гидратации
По правилу Марковникова – присоединение к непредельным углеводородам несимметричного строения соединений с общей формулой НХ - атом водорода присоединяется к наиболее гидрогенизированому атому углерода ,если заместитель ЭД. При антимарковниковском присоединении атом водорода присоединяется к наименее гидрогенизированному, если заместитель ЭА.
Реакции электрофильного замещения в ароматических системах имеют свои особенности. Первая особенность состоит в том, что для взаимодействия с термодинамически устойчивой ароматической системой требуются сильные электрофилы, которые, как правило, генерируются с помощью катализаторов.
Механизм реакции SE 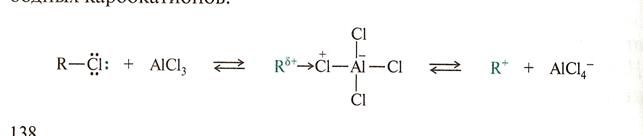

ОРИЕНТИРУЮЩЕЕ ВЛИЯНИЕ
ЗАМЕСТИТЕЛЕЙ
Если в ароматическом ядре находится какой-либо заместитель, то он обязательно оказывает влияние на распределение электронной плотности кольца. ЭД – заместители (ориентанты 1-го ряда) СН3, ОН, ОR, NН2, NR2 – облегчают замещение по сравнению с не замещенным бензолом и направляют входящую группу в орто- и пара- положение. Если ЭД заместители сильные , то не требуется катализатор эти реакции протекают в 3 стадии.
ЭА – заместители ( ориентанты II-го рода) затрудняют реакции электрофильного замещения по сравнению с не замещенным бензолом. Реакции SЕ идет в более жестких условиях, входящая группа вступает в мета положение. К заместителям II рода относятся:
СООН, SО3Н, СНО, галогены и др.
Реакции SЕ характерны также для гетероциклических углеводородов. Пиррол, фуран, тиофен и их производные относятся к π- избыточным системам и достаточно легко вступает в реакции SЕ. Они легко галогенируются, алкилируются, ацилируются, сульфируются, нитрируются. При выборе реагентов необходимо учитывать их не стабильность в сильнокислотной среде т.е ацидофобность.
Пиридин и другие гетероциклические системы с пиридиновым атомом азота ,являются π –не достаточными системами, они гораздо труднее вступают в реакции SЕ, при этом входящий электрофил занимает β-положение по отношению к атому азота. 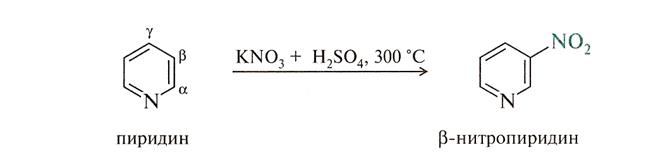
Кислотные и основные свойства органических соединений
Важнейшими аспектами реакционной способности органических соединений являются кислотно-основные свойства органических соединений.
Кислотность и основность также важные понятия, определяющие многие функциональные физико-химические и биологические свойства органических соединений. Кислотный и основной катализ является одной из наиболее распространенных ферментативных реакций. Слабые кислоты и основания – обычные компоненты биологических систем, играющие важную роль в метаболизме и его регуляции.
В органической химии существует несколько концепций кислот и оснований. Общепринятая в неорганической и органической химии теория кислот и оснований Бренстеда. Согласно Бренстеду, кислоты представляют вещества, способные отдать протон, а основания – вещества, способные присоединить протон. 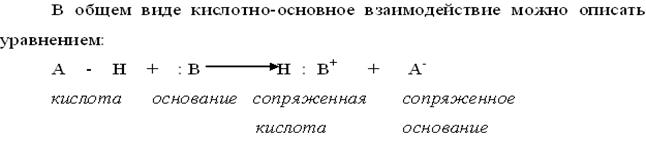
Кислотность по Бренстеду
В принципе, большинство органических соединений можно рассматривать как кислоты, поскольку в органических соединениях Н связан с С, N O S
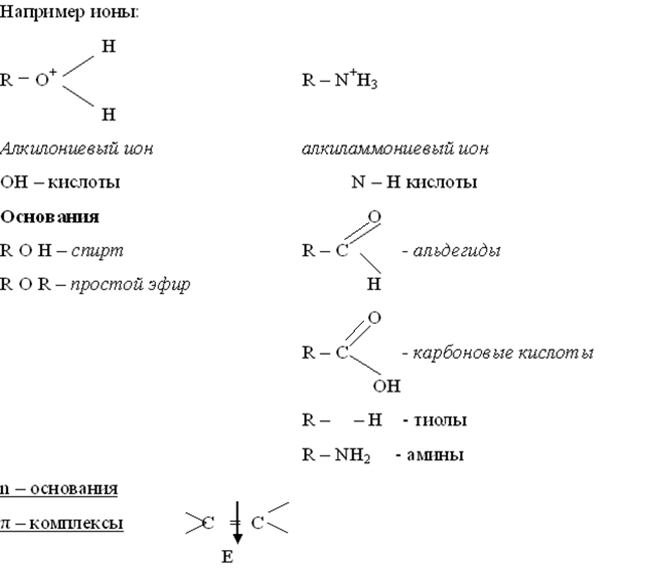
Органические кислоты соответственно делятся на С – Н, N – Н, О – Н, S-Н – кислоты. 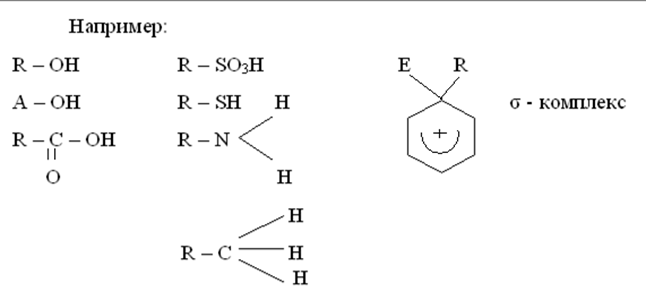
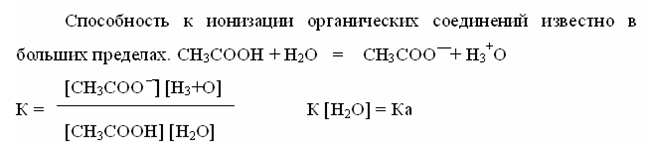
Кислотность оценивается в виде Ка или - lg Ка = рКа, чем меньше рКа, тем сильнее кислота.
Количественная оценка кислотности органических соединений определена далеко не у всех органических веществ. Поэтому важно выработать умение проводить качественную оценку кислотных свойств различных кислотных центров. Для этого используют общий методический подход.
Сила кислоты определяется стабильностью аниона( сопряженного основания). Чем стабильнее анион, тем сильнее кислота.
Стабильность аниона определяется совокупностью ряда факторов:
электроотрицательностью и поляризуемостью элемента в кислотном центре.
степенью делокализации отрицательного заряда в анионе.
характером связанного с кислотным центром радикала.
сольватационными эффектами (влияние растворителя)
Рассмотрим последовательно роль всех этих факторов:
Влияние электроотрицательности элементов
Чем более электроотрицателен элемент, тем более делокализован заряд и тем стабильнее анион, тем сильнее кислота.
С (2,5) N (3,0) О(3.5) S (2,5)
Поэтому кислотность изменяется в ряду СН< NН < ОН<SH
Для SH – кислот преобладает другой фактор – поляризуемость.
Атом серы больше по размеру и имеет вакантные d – орбитали. следовательно, отрицательный заряд способен делокализоваться в большом объеме, что приводит к большей стабильности аниона.
Тиолы, как более сильные кислоты, реагируют с щелочами, а также с оксидами и солями тяжелых металлов, тогда, как спирты (слабые кислоты) способны реагировать только с активными металлами 
Относительно высокая кислотность толов используется в медицине, в химии лекарственных средств. Например: 
Применяют при отравлениях As, Hg, Cr, Bi, действие которых обусловлен связыванием металлов и выведением их из организма. Например: 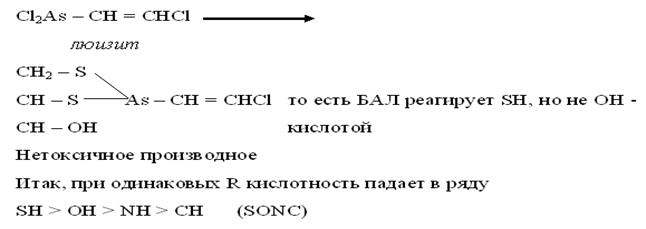
При оценке кислотности соединений с одинаковым атомом в кислотном центре определяющим фактором является делокализация отрицательного заряда в анионе. Стабильность аниона значительно повышается с появлением возможности делокализации отрицательного заряда по системе сопряженных связей. Значительное увеличение кислотности в фенолах, по сравнению со спиртами объясняется возможностью делокализации в ионах по сравнению с молекулой. 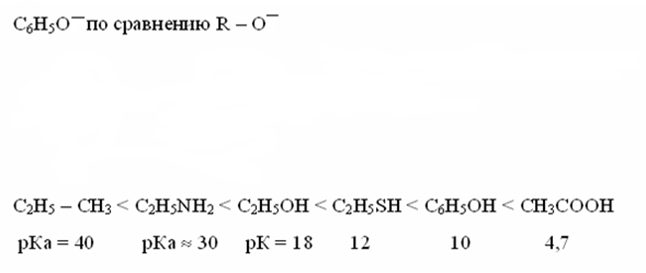
Высокая кислотность карбоновых кислот обусловлена резонансной стабильностью карбоксилат аниона 
Делокализация заряда способствует наличие электроноакцепторных заместителей (ЭА) они стабилизируют анионы, тем самым увеличивают кислотность. Например, введение в молекулу ЭА заместителя 
Влияние заместителя и растворителя
a - оксикислоты более сильные кислоты, чем соответствующие карбоновые кислоты.
ЭД – заместители наоборот понижают кислотность. Растворители оказывают большее влияние на стабилизацию аниона, как правило лучше сольватируются небольшие ионы с низкой степенью делокализации заряда.
Влияние сольватации можно проследить например в ряду: 
Если атом в кислотном центре несет положительный заряд это приводит к усилению кислотных свойств.
Вопрос к аудитории: какая кислота – уксусная или пальмитиновая С15Н31СООН – должна иметь меньшее значение рКа?
Если атом в кислотном центре несет положительных заряд, это приводит к усилению кислотных свойств. 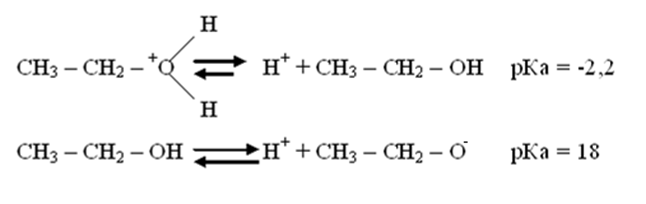
Можно отметить сильную СН – кислотность σ – комплекса, образующегося в реакции электрофильного замещения. 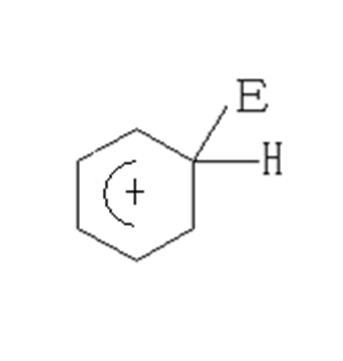
Основность по Бренстеду
Для того, чтобы образовать связь с протоном, необходимо не поделенная электронная пару у гетероатома,
либо быть анионами. Существуют п-основания и
π-основания, где центром основности являются
электроны локализованной π-связи или π-электроны сопряженной системы (π-компоненты) 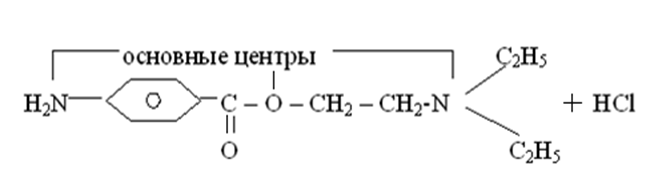
Сила основания зависит от тех же факторов, что и кислотность, но влияние их противоположно. Чем больше электроотрицательность атома, тем прочнее он удерживает неподеленную пару электронов, и тем менее доступна она для связи с протоном. Тогда в целом сила n-оснований с одинаковым заместителем изменяется в ряду:
N > O > S
Наибольшую основность из органических соединений проявляют амины и спирты:
Соли органических соединений с минеральными кислотами хорошо растворимы. Многие лекарственные средства используют в виде солей.
Кислотно-основной центр в одной молекуле(амфотерность) 

Водородные связи как кислотно-основное взаимодействие
Для всех α – аминокислот является преобладание катионных форм в сильнокислых и анионных в сильнощелочных средах.
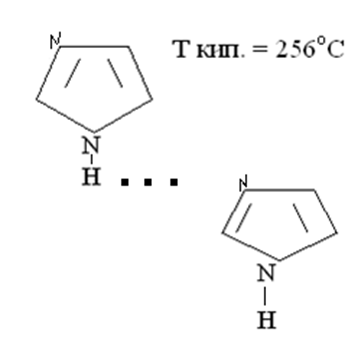
Наличие слабых кислотных и основных центров приводит к слабым взаимодействиям – водородным связям. Например: имидазол при небольшой молекулярной массе имеет высокую температуру кипения за счет наличия водородных связей.

Дж. Льюисом предложена более общая теория кислот и оснований, определяющаяся на строении электронных оболочек.
Кислотами по Льюису могут быть атом, молекула или катион, обладающие вакантной орбиталью, способное принимать пару электронов с образованием связи.
Представителями кислот Льюиса служат галогениды элементов II и III групп периодической системы Д.И. Менделеева.
Основания Льюиса атом, молекула или анион способный предоставлять пару электронов.
К основаниям Льюиса относятся амины, спирты, простые эфиры, тиолы, тиоэфиры и содержащие π-связи соединения.
Например, приведенное ниже взаимодействие можно представить как взаимодействие кислот и оснований Льюиса 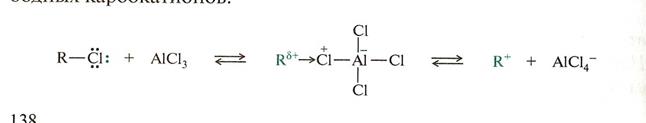
Важным следствие теории Льюиса является то, что любое органическое вещество можно представить как кислотно-основной комплекс.
В органических соединениях внутримолекулярные водородные связи возникают значительно реже, чем межмолекулярные, но также имеют место в биоорганических соединениях и их можно рассматривать как кислотно-основные взаимодействия. 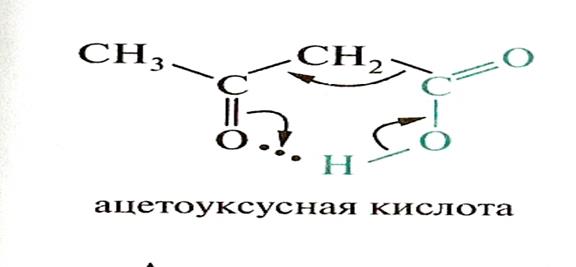
Понятие «жесткие» и «мягкие» не тождественны сильным и слабым кислотам и основаниям. Это независимые две характеристики. Суть ЖКМО состоит в том, что жесткие кислоты реагируют с жесткими основаниями и мягкие кислоты реагируют с мягкими основаниями. 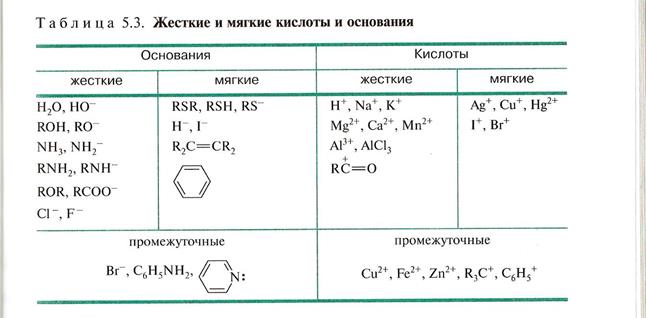
ЖМКО
В соответствии с принципом жестких и мягких кислот и оснований (ЖМКО) Пирсона кислоты Льюиса делятся на жесткие и мягкие. Жесткие кислоты- акцепторные атомы с малым размером ,большим положительным зарядом, большой электроотрицательностью и низкой поляризуемостью.
Мягкие кислоты- акцепторные атомы большого размера с малым положительным зарядом, с небольшой электроотрицательностью и высокой поляризуемостью.
Суть ЖКМО состоит в том, что жесткие кислоты реагируют с жесткими основаниями и мягкие кислоты реагируют с мягкими основаниями . Например: 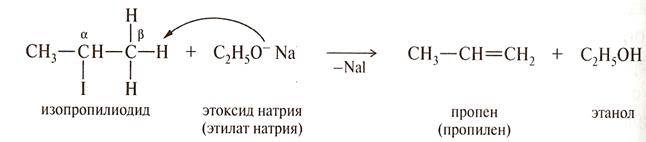

Окисление и восстановление органических соединений
Окислительно-восстановительные реакции занимают важнейшее значение для процессов жизнедеятельности. С их помощью организм удовлетворяет свои энергетические потребности, поскольку при окислении органических веществ происходит высвобождение энергии.
С другой стороны эти реакции служат для превращения пищи в компоненты клетки. Реакции окисления способствуют детоксикации и выведению лекарственных средств из организма.
Окисление – процесс удаления водорода с образованием кратной связи или новых более полярных связей
Восстановление – процесс обратный окислению.
Окисление органических субстратов протекает тем легче, чем сильнее его тенденция к отдаче электронов.
Окисление и восстановление необходимо рассматривать по отношению к определенным классам соединений.
Окисление С – Н связей (алканов и алкилов)
При полном сгорании алканов образуется СО2 и Н2О при этом выделяется тепло. Другие пути их окисления и восстановления можно представить следующими схемами: 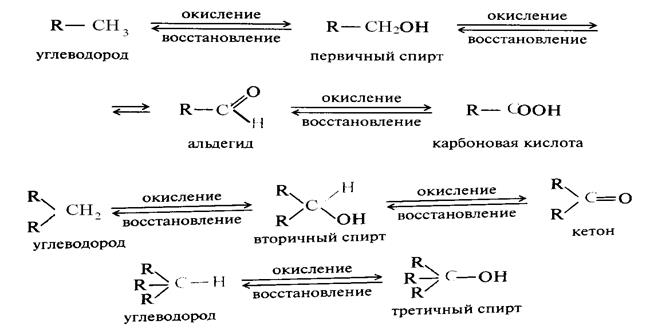
Окисление насыщенных углеводородов протекает в жестких условиях (хромовая смесь горячая) более мягкие окислители не действуют на них. Промежуточными продуктами окисления являются спирты, альдегиды, кетоны, кислоты.
Гидропероксиды R – О – ОН важнейшие промежуточные продукты окисления С – Н связей в мягких условиях, в частности in vivo
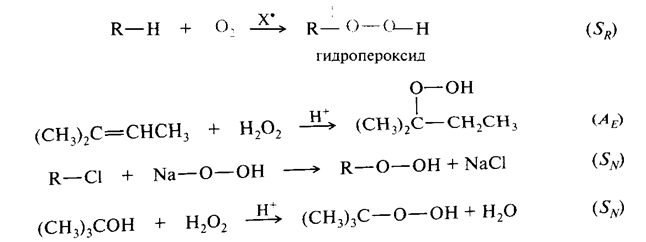
Важной реакцией окисления С – Н связей в условиях организма является ферментативное гидроксилирование.
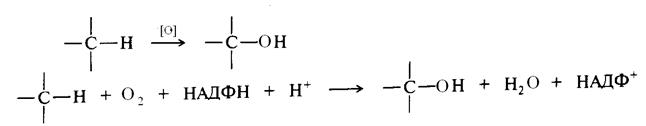
Примером может быть получение спиртов при окислении пищи . За счет молекулярного кислорода и его активных форм . осуществляется в in vivo.
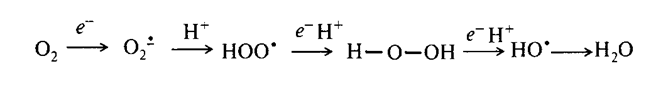 Перекись водорода может служить в организме гидроксилирующим агентом.
Перекись водорода может служить в организме гидроксилирующим агентом.
Избыток перекиси должен разлагаться с помощью каталазы на воду и кислород.
Окисление и восстановление алкенов можно представить следующими превращениями: 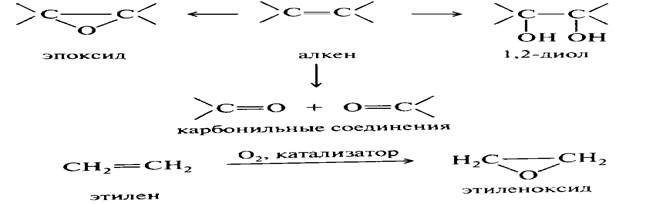
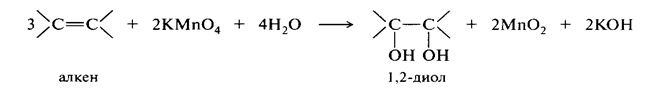
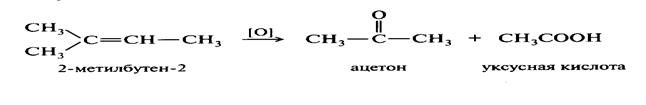
Восстановление алкенов
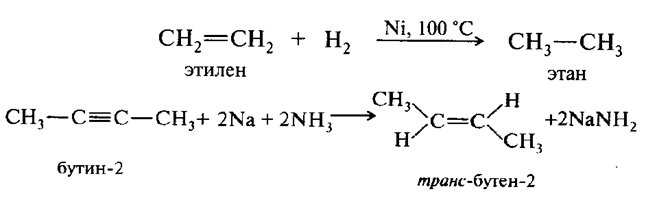
Окисление и восстановление ароматических углеводородов
Бензол чрезвычайно тяжело окисляется даже в жестких условиях по схеме:
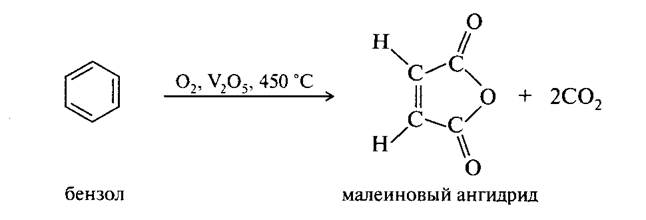
Способность к окислению заметно увеличивается от бензола к нафталину и далее к антрацену.
ЭД- заместители облегчают окисление ароматических соединений. ЭА – затрудняют окисление. Восстановление бензола.
С6Н6 + 3Н2 
Ферментативное гидроксилирование ароматических соединений
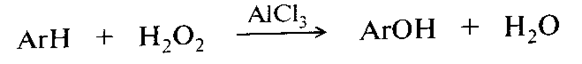

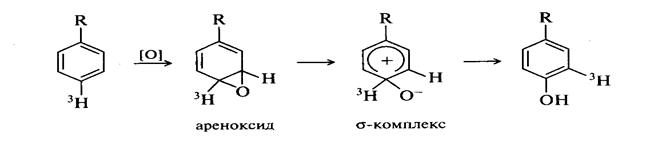
Окисление спиртов
По сравнению с углеводородами, окисление спиртов осуществляется в более мягких условиях 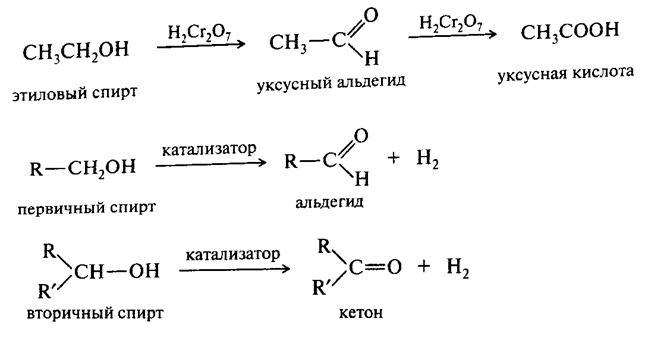
Важнейшей реакцией диолов в условиях организма является превращения в системе хинон-гидрохинон 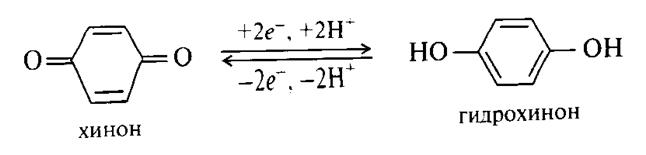
Перенос электронов от субстрата к кислороду осуществляется в метахондриях.
Окисление и восстановление альдегидов и кетонов
Один из наиболее легко окисляющийся классов органических соединений 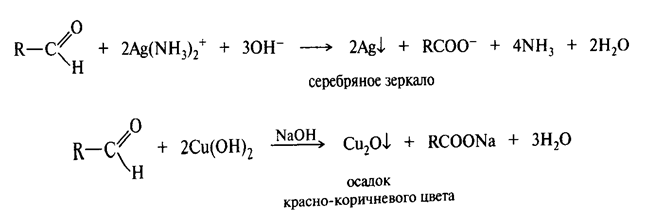
2Н2С = О + Н2О СН3ОН + НСООН особенно легко протекает на свету
Окисление азотсодержащих соединений
Амины окисляются достаточно легко конечными продуктами окисления являются нитросоединения 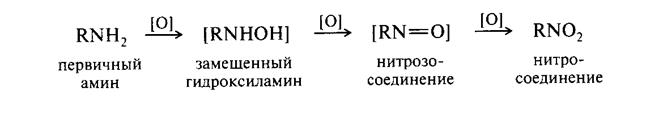
Исчерпывающее восстановление азотсодержащих веществ приводит к образованию аминов. 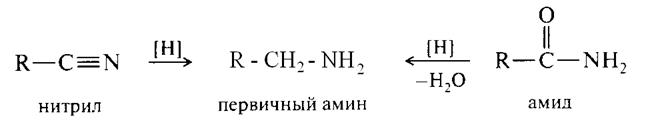
Окисление аминов в in vivo 
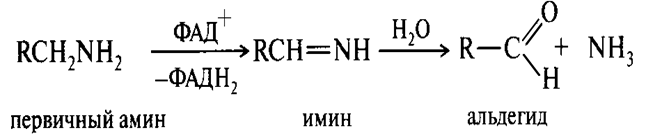
Окисление и восстановление тиолов 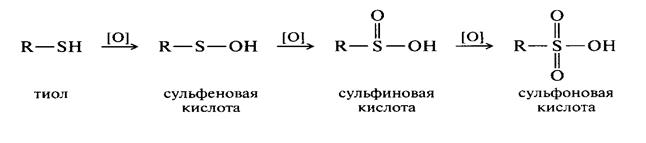
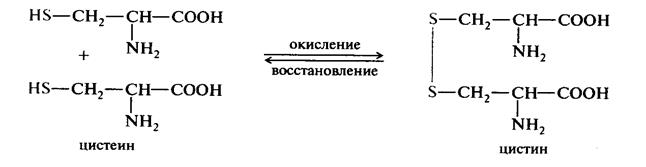
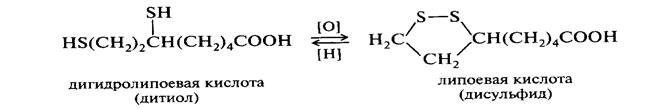
Cравнительная характеристика О-В свойств органических соединений.
Наиболее легко окисляются тиолы и 2-х-атомные фенолы. Достаточно легко окисляются альдегиды. Труднее окисляются спирты, причем первичные легче, чем вторичные, третичные . Кетоны устойчивы к окислению или окисляются с расщеплением молекулы.
Алкины окисляются легко даже при комнатной температуре.
Наиболее трудно окисляются соединения, содержащие атомы углерода в Sр3-гибридизированом состоянии, то есть насыщенные фрагменты молекул.
ЭД – заместители облегчают окисление
ЭА – затрудняют окисление.
Специфические свойства поли- и гетерофункциональных соединений.
План лекции
Поли- и гетерофункциональность, как фактор повышающий реакционную способность органических соединений.
Специфические свойства поли- и гетерофункциональных соединений:
амфотерность образование внутримолекулярных солей.
внутримолекулярная циклизация γ, δ, ε – гетерофункциональных соединений.
межмолекулярная циклизация (лактиды и декетопипирозины)
хелатообразование.
реакции элиминирования бета – гетерофункциональных
соединений.
таутомерия кето–енольная. Фосфоенолпируват, как
макроэргическое соединение.
декарбоксилирование.
стереоизомерия
Поли-и гетерофункциональность, как причина появления специфических свойств у гидрокси-, амино- и оксокислот.
Наличие в молекуле нескольких одинаковых или разных функциональных групп составляет характерную черту биологически важных органических соединений. В молекуле может быть две и более гидроксильных групп, аминогрупп , карбоксильных групп. Например: 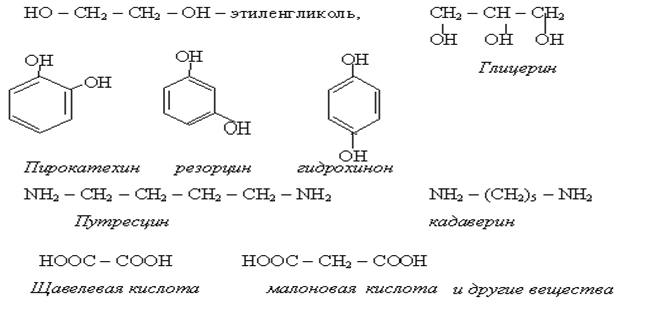
Важную группу веществ участников жизнедеятельности составляют гетерофункциональные соединения, имеющие попарное сочетание разных функциональных групп. Например:
В алифатических соединениях все приведённые функциональные группы проявляют ЭА характер. За счёт влияния друг на друга у них взаимно усиливается реакционная способность. Например, в оксокислотах электрофильность усиливается каждого из двух карбонильных атомов углерода под влиянием -J другой функциональной группы, что ведёт к более легкому восприятию атаки нуклеофильными реагентами.
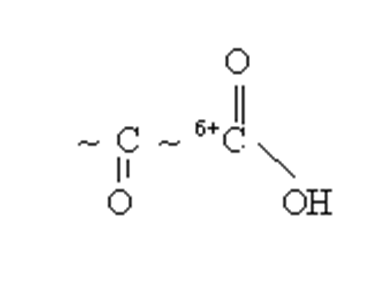
Поскольку I эффект затухает через 3–4 связи, то важным обстоятельством является близость расположения функциональных групп в углеводородной цепи. Гетерофункциональные группы могут находится у одного и того же атома углерода (α – расположение), или у разных атомов углерода как соседних(β расположение), так и более удалённых друг от друга (γ, дельта, эпсилон) расположения.
Каждая гетерофункциональная группа сохраняет собственную реакционную способность, точнее гетерофункциональные соединения вступают как бы в «двойное» число химических реакций. При достаточном близком взаимном расположении гетерофункциональных групп происходит взаимное усиление реакционной способности каждой из них.
При одновременном присутствии в молекуле кислотной и основной групп, соединение становятся амфотерным.
Например: аминокислоты.
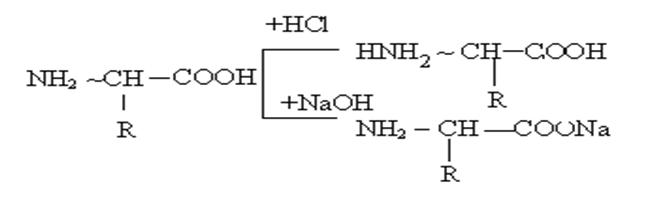
Взаимодействие гетерофункциональных групп
В молекуле герофункциональных соединений могут содержатся группы, способные к взаимодействию друг с другом. Например, в амфотерных соединениях, как в α- аминокислотах, возможно образование внутренних солей. 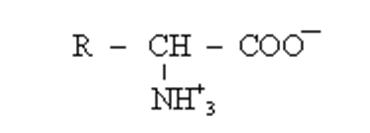
По этому все α – аминокислоты встречаются в виде биополярных ионов и хорошо растворимы в воде.
Кроме кислотно–основных взаимодействий становятся возможны и другие виды химических реакций. Например, реакции SN у SP2 гибрид атома углерода в карбонильной группе за счёт взаимодействия со спиртовой группой образование сложных эфиров, карбоксильной группы с аминогруппой (образование амидов).
В зависимости от взаимного расположения функциональных групп эти реакции могут протекать как внутри одной молекулы (внутримолекулярные), так и между молекулами (межмолекулярные).
Поскольку в результате реакции образуется циклические амиды, сложные эфиры. то определяющим фактором становится термодинамическая устойчивость циклов. В связи с этим конечный продукт, как правило, содержит шестичленный или пятичленный циклы.
Чтобы при внутримолекулярном взаимодействии образовался в пяти или шестичленный сложноэфирный (амидный) цикл, гетерофункциональное соединение должно иметь в молекуле гамма или сигма расположение. Тогда в кл
Дата добавления: 2016-10-07; просмотров: 5663;
Поиск по сайту
Узнать еще
- Аналитическая химия и химический анализ. Предмет и задачи аналитической химии. Классификация методов химического анализа.
- БИОЛОГИЧЕСКАЯ ХИМИЯ
- БИОХИМИЯ ЗУБНОГО НАЛЕТА. ЗУБНОЙ НАЛЕТ И КАРИЕС.
- Биохимия осложнений сахарного диабет.
- БИОХИМИЯ СОЕДИНИТЕЛЬНОЙ ТКАНИ
- ГЕОХИМИЯ КАК НАУКА. ПРЕДМЕТ И МЕТОД ГЕОХИМИИ
- Естественноисторический подход и эволюционная химия

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
