Валентность, степень окислении, координационное число
ТИПЫ И ОСОБЕННОСТИ ХИМИЧЕСКИХ СВЯЗЕЙ
Валентное связывание
Ионная связь
Ионная связь (ИС) относится к связям валентного типа, поскольку образование ионных соединений из элементов протекает с изменением их валентного состояния (здесь не рассматриваются реакции между ионными соединениями, т.е. вторичные реакции).
ИС возникает при взаимодействии элементов с большой разницей в их электроотрицательностях (ЭО). Под понятием ЭО его творец – Лайнус Полинг - понимал “способность атома в молекуле притягивать электроны”, иными словами ЭО элемента есть сила, удерживающая электроны у данного атома в молекуле. Последнее добавление указывает на зависимость величины ЭО данного атома от природы атомов его окружающих.
Безразмерный коэффициент c, характеризующий ЭО, выведен из термохимических данных с использованием понятия энергии ионного резонанса (ЭИР). По мнению Полинга ЭИР возникает при образовании гетероатомных молекул из гомоатомных и упрочняет первые. Например, связь F-Cl более прочна, чем связи в Cl-Cl и F-F. Отсюда ЭИР равна разнице между значением рассчитанной энергии ковалентной связи и экспериментальным значением общей энергии связи) и ее величина является характеристикой ЭО. Для наиболее электроотрицательного атома фтора c=4, а для наиболее электроположительного атома франция c=0.7 (для атома Н принято значение 2.2 и относительно него выводятся коэффициенты для всех остальных атомов). Понятно, что все эти величины относительны и условны
В шкале Малликена ЭО определяется как величина пропорциональная сумме энергий ионизации атома и сродства к электрону и выражается в ккал/моль, хотя и в этом случае отношение cF/cLi все равно близко к 4.
В шкале Оллреда-Рохова, наиболее популярной у химиков, ЭО определяется как электростатическая сила, действующая между ядром и валентными электронами.
Для расчета коэффициентов ЭО используется величины эффективных ядерных зарядов Z* (ЭЯЗ – часть заряда ядра, затраченного для притяжения валентных электронов Z*=Z-S, где Z- общий заряд ядра, а S –константа экранирования ядра внутренними электронами), вычисляемых с помощью правил Слэтера, и значений ковалентных радиусов атомов.
Следует еще раз подчеркнуть, что величина ЭО не постоянна и зависит от валентного состояния атома, т.е. его заряда, и характера его окружения, т.е. от типа гибридизации его валентных орбиталей и природы заместителей.
В идеальном случае возникновение ИС означает образование в ходе взаимодействия исходных веществ ионов, т.е. частиц с разделенным зарядом, реально существующих в продуктах реакции. Существование химических веществ с подобным типом связи – пожалуй, первый в химии хорошо установленный экспериментальный факт. Это доказательство ведет свое начало с теории электролитической диссоциации (С. Аррениус). В настоящий момент для нашего рассмотрения не имеет значения другие хорошо установленные факты. В частности, соединений с чисто ионной связью в природе не существует, поскольку во всех случаях, даже в фторидах щелочных металлов, химическая связь в ионных кристаллах содержит в том или ином количестве ковалентную составляющую, доля которой в связи зависит от поляризующей способности катиона (три правила Фаянса): 1- она тем больше, чем больше заряд катиона и меньше его размер, 2- чем выше заряд и размер аниона, 3- чем дальше строение электронной оболочки катиона от строения оболочки ближайшего инертного газа (последняя эффективно экранирует заряд ядра и препятствует поляризации аниона), и от того, что характер электронного распределения в молекуле может меняться в зависимости от лигандного окружения (цвиттер-ионы) или от электронного и колебательного состояния молекулы в данный момент.
Электростатическая модель позволяет успешно предсказывать энергию ионной решетки, что также подтверждает факт существования ионов и дает возможность определить из структурных данных, как их радиусы, так и факторы, влияющие на их размер. Знание радиусов атомов и ионов в современной химии весьма велико, поскольку позволяет предсказывать структурные типы вновь полученных соединений, проводить обсуждение причин отклонения их химического поведения от ожидаемого или объяснить появление других необычных свойств вещества. Помимо этого сама шкала ЭО по Оллреду-Рохову базируется на знании ковалентных радиусов элементов.
В веществах с ИС каждый ион окружен противоионом. Например, в CsCl катион Cs+ окружен 8 анионами Cl-, и наоборот – анион Cl- окружен 8 катионами Cs+, т.е. мы имеем дело с ненаправленной и ненасыщаемой связью (см. ниже) относительно каждого иона. Именно это свойство ИС обуславливает возможность получения кристаллов ионных соединений с переменным составом. Как правило, эти вещества имеют относительно небольшую область гомогенности, не очень сильно нарушающую стехиометрию в его составе. Например, в NaCl область гомогенности не превышает 0.1 ат. % (в ковалентных она в силу насыщенности и направленности связи еще меньше и в самых крайних случаях не превышает 0.01 ат. %). В то же время известны преимущественно ионные соединения с очень широкой областью гомогенности, которые являются составной частью класса нестехиометрических веществ или бертоллидов, и в которых не соблюдается закон кратных отношения Дальтона, точнее он соблюдается, но виртуально.
В наибольшей степени нестехиометричность типична для соединений с металлическим типом связи, но известны десятки и сотни веществ с смешанным типом связи (ионно-металлическая), к которым, как правило, относятся оксиды, сульфиды, теллуриды и селениды и для которых также характерно это свойство. Так, в FeS количество железа может меняться в пределах от 35 до теоретических 50 ат. %. Однако получить сульфид железа стехиометрического состава при обычных условиях оказывается невозможным, поскольку на диаграмме состояния он (состав) располагается за пределами области гомогенности. В TiO атомная доля кислорода колеблется в пределах 0.6 - 1.4 и сильно зависит от способа получения вещества.
Рассмотренные примеры, в целом, нетипичны для соединений ионного типа, поскольку по величинам отклонения от стехиометрии они приближаются к интерметаллическим соединениям и даже металлидам – чемпионам среди веществ с нарушенной стехиометрией. Обычно эти отклонения в ионных соединениях существенно меньше, тем не менее, точный стехиометрический состав для них скорее исключение, чем правило. Электронейтральность в этих соединениях поддерживается самыми разнообразными способами – путем создания компенсирующих дефектов, например, появление в структуре дополнительного электрона компенсируется появлением в решетке дырки или путем образования связей М-М или кластеров.
Среди неорганических соединений вещества нестехиометрического состава привлекают наибольшее внимание исследователей, поскольку любая нестехиометрия заметно влияет на самые различные физические, химические и механические свойства вещества, например на электро- и фотопроводимость, прочность, адсорбцию и именно поэтому направленный синтез подобных веществ является предметом современного фундаментального материаловедения. Яркий пример последних десятилетий – высокотемпературные меднооксидные сверхпроводники, в которых сверхпроводящие свойства определяются кислородной нестехиометрией.
Разделение заряда в рамках одной ковалентно связанной молекулы приводит к образованию цвиттер-ионов или биполярных ионов. Подобную структуру имеют, например, аминокислоты, существующие в виде внутренних солей – бетаинов –(СH3)3N+CH2COO- « (СH3)2NCH2COOCH3, мезоионные соединения – пяти или шестичленные азотсодержащие циклы, в которых имеется один или несколько внекольцевых азот-, кислород- или серосодержащих заместителей, илиды общей формулы RnZ+ - C¯ < « RnZ=C<, где Z- N, P, As, Sb, Bi, O, Se, Te, Cl, Br, I, некоторые металлорганические соединения, например Сp(Ph)Zr+[C5H4CH2B-(C6F5)2]C6F5. Последние вещества играют важную роль в металлокомплексном катализе как активные центры в т.н. ценовых катализаторов реакций полимеризации и олигомеризации алкенов.
Следует отметить способность ряда чисто органических соединений образовывать в определенных условиях, например при использовании растворителей с высокой диэлектрической проницаемостью, карбкатионы и карбанионы, что переводит достаточно медленные реакции ковалентных органических соединений в быстрые ионные.
Ковалентная связь
Наиболее распространенный тип связи в органических, координационных и металлорганических веществах и в меньшей степени в неорганических соединениях. Это, как правило, самый сильный по энергетике тип межатомных взаимодействий, протекающий с изменением валентного состояния атомов и характеризуемый перераспределением и сгущением электронной плотности между взаимодействующими атомами по сравнению с свободными атомами. В рамках классической теории Льюиса ковалентное связывание объясняется образованием электронных пар общих для связываемых атомов, что приводит к достраиванию их электронных оболочек до замкнутых оболочек инертных газов. Теория Льюиса является феноменологической теорией, поскольку она, объясняя следствие, не объясняет причин возникновения химической связи. Эти причины раскрываются в методах валентных связей и молекулярных орбиталей, которые рассматриваются в самых различных курсах химии.
В отличие от ИС ковалентная связь является направленной,поскольку электронная плотность МО, сосредоточена в ближайшем и ограниченном объеме пространстве, взаимодействующих атомов, определяемом направлением перекрывания АО (понятие гибридизация атомных орбиталей и причина близкого строения родственных соединений, точнее их фрагментов). И насыщаемой,поскольку в ограниченном объеме пространства вокруг каждого атома возможно размещение ограниченного числа электронных пар при условии соблюдения принципа Паули. Напротив, ионная связь, как отмечалось выше, ненаправлена и ненасыщаема, поскольку в пространстве между ионами возможно размещение и наложение друг на друга нескольких зарядов (во всяком случае таких запретов нет) и существует дальнодействие зарядов, распространяющееся на вторую, третью и т.д координационные сферы c силой, соответствующей закону Кулона (qq’/r2). Вследствие таких различий ковалентно связанные соединения стехиометричны, а дефектность кристаллов имеет совершенно иную природу, чем в ионных веществах или металлидах, и иные следствия.
Другая особенность большинства из ковалентных соединений заключается в том, что, несмотря на прочность связи между атомами, связь между молекулами очень слаба и обеспечивается межмолекулярным (вандерваальсовым) взаимодействием. Это приводит к низким температурам плавления и высокой летучести большинства ковалентно связанных соединений. Причина прочности и высокой температурной стойкости некоторых, безусловно, ковалентных веществ, например алмаза или кубического нитрида бора, заключается в том, что их кристаллы построены из одной молекулы, т.е. в них просто отсутствует межмолекулярное связывание.
Общее понятие “ковалентная связь” включает несколько ее вариантов – “классические” ковалентные двухцентровые связи, полярные, многоцентровые. В последние годы в этом же разделе рассматриваются донорно-акцепторные, агостические и дативные связи, хотя это, видимо, не совсем правомерно, т.к. они образуются без изменения валентного состояния атомов, а последние вообще возникают как следствие образования «нормальной» ковалентой связи. Но, с одной стороны, методологически все они описываются одними и теми же уравнениями в методе МО, а, с другой, отражают особенности ковалентного связывания между неметаллами, металлами и неметаллами и их различными комбинациями в гомо- и гетероядерных структурах. Отметим некоторые из них.
Сдвиг электронной плотности к наиболее электроотрицательному атому, в конце концов, приводит к поляризации, ионизации связи, а затем и к образованию соединения, построенного по ионному типу. Отсюда - ионная связь – крайний случай ковалентной, а другой крайний случай - “классическая” ковалентная двухцентровая s-связь, обусловленная перекрыванием s-орбиталей и наблюдаемая, например в молекуле водорода. При подобном связывании электронная пара находится точно по центру линии водород-водород. Перекрывание р-орбиталей, расположенных вне линии соединяющей атомы, приводит к образованию p-связей и соединений с кратными связями. В основном это многочисленные органические и металлорганические соединения, но среди них есть и неорганические вещества, например диазот или окись углерода с тройными связями между атомами. Органические соединения с кратными связями относятся к предмету органической химии и, в общем, их рассмотрение, с одной стороны, выходит за рамки данной лекции, а, с другой, уже достаточно тривиально. В то же время образование соединений с кратными связями М-М еще сравнительно недавно было необычно и ново.
Кратные связи метал-металл, наблюдаемые в кластерных соединениях невысокой нуклеарности, а обычно в биядерных комплексах, могут быть двойными, тройными и даже четверными, что приводит к существенному сокращению межатомного расстояния М-М против наблюдаемого в компактном металле. Впервые эти связи были открыты в биядерных хлоридах рения, находящегося в степени окисления +3, прежде всего в Re2Cl8-2, имеющем кратность связи равную 4 и наиболее «старую» историю существования (рис.1).
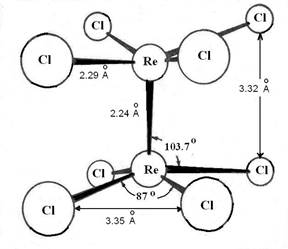
Рис.1. Строение октахлордиренат-иона.
В этом анионе длина связи Re-Re=2.24 Å, в то время как в металле она равна 2.75 Å. Теория, объясняющая образование кратных связей этого типа, относительна проста и понятна и впервые разработана одним из первооткрывателей соединений этого типа Ф. Коттоном в 1964 году (хотя впервые соединение с этим анионом и первые, но не вполне правильно осмысленные и объясненные структурные данные получены советскими учеными в 1960 г.).
При сближении двух атомов металлов между парами d-орбиталей в силу свойств их симметрии могут возникнуть только пять ненулевых перекрываний dz2 , dyz, dxz, dxy и dx2-y2 (рис. 2 и рис. 3а). Положительное перекрывание двух dz2-орбиталей приводит к образованию связывающей σ-орбитали. Это первая и наиболее прочная составляющая связи Re-Re. Каждое из положительных перекрываний dxz и dyz-орбиталей, приводит к двум π-составляющим связи Re-Re, расположенным в плоскостях xz и yz. Оставшиеся dxy и dx2-y2-орбитали должны участвовать в образовании наиболее слабой δ-связи. Поскольку,
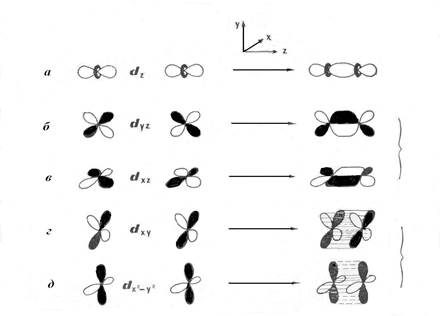
Рис.2. Пять ненулевых перекрываний d-орбиталей двух атомов металла.
однако, dx2-y2-орбитали расположены так, что ориентированы к лигандам (в анионе Re2Cl8-2 к атомам хлора (рис. 1, рис 3), они участвуют в образовании σ-связи металл-лиганд и из рассмотрения исключаются. Таким образом, четвертая компонента связи М-М обеспечивается перекрыванием двух dxy-орбиталей, при этом такое перекрывание становится возможным только при расположении орбиталей строго друг под другом (рис. 3 в). Именно поэтому хлорные лиганды в Re2Cl8-2 расположены в энергетически невыгодной заслоненной конформации. Поскольку атом рения в анионе имеет формальную степень окисления +3, то на каждом атоме остается 7-3=4 валентных электрона (d4 электронная конфигурация). Эти восемь электронов от двух атомов Re заполняют связывающие орбитали, давая σ2π4δ2 конфигурацию. При этом разрыхляющие орбитали остаются вакантными. Поскольку порядок связи определяется как разность между числом связывающих и разрыхляющих электронов, деленная на 2, порядок связи Re-Re в Re2Cl8-2 будет равным 4.
Очевидные пути снижения кратности до 3 – удаление двух электронов с δ-орбитали, что приводит к реализации σ2π4 конфигурации, или прибавление двух электронов на разрыхляющую δ*-орбиталь с образованием σ2π4δ2δ*2 конфигурации. Удаление или приобретение одного электрона дает порядок связи 3,5 с конфигурациями σ2π4δ2δ* или σ2π4δ. Аналогичным образом, заполняя разрыхляющие орбитали или снимая электроны с связывающих орбиталей, можно получить весь набор кратностей вплоть до 1.
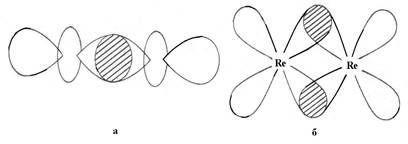
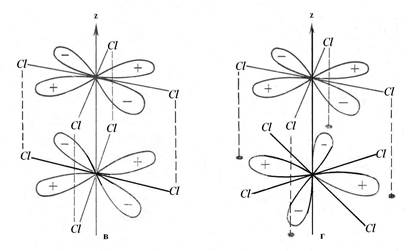
Рис. 3. Образование связи между атомами рения: а- σ-связь; б- π связь; в- δ связь, образующаяся при расположении атомов хлора друг над другом; г- отсутствие перекрывания dxy орбиталей при скошенном расположении атомов хлора.
Несмотря на высокий порядок связи и короткие межатомные контакты энергии кратных связей М-М, образованных за счет перекрывания s, p и d орбиталей от d-электронов, существенно ниже энергий кратных связей, образованных неметаллами за счет перекрывания р-орбиталей. Заметно отличаются и их химические свойства, например, в таких реакциях как реакции присоединения. Для органических соединений с ненасыщенными связями эта реакция характеристична. Для кластеров с кратными связями присоединение также может идти по этому пути (например, в триосмиевом карбониле с одной двойной связью), но может реализоваться совершенно иной путь преобразования кластерной молекулы, например, связанный с реакциями замещения лигандов или расщепления металлического каркаса.
Возможно ли существование соединений с более высокой кратностью связи? Ответ на этот вопрос вызывает споры до настоящего времени. Большая часть химиков –теоретиков считает, что кратность связи М-М равная 4 предельна. Меньшая полагает, что это не так и рассматривает молекулы Мо2 как вероятные, а W2 уже как абсолютно надежные примеры соединений с кратностью связи равной 6. Но эти предположения не подтверждены экспериментом. В то же время пока в структурно охарактеризованных комплексах хрома+1 состава ArCrCrAr и молибдена (рис. 4 a) предполагают кратность связи равную 5. Аналогичная кратность возможна и в комплексе ArUUAr.
Еще 10-15 лет тому назад считалось, что кратные связи М-М могут существовать только у элементов переходных периодов, поскольку согласно теоретическим представлениям для их образования необходимо участие орбиталей с квантовым числом углового момента равным 2 (d-орбитали) или больше (f,g-орбитали). Но в конце XX и начале XXI веков и этот запрет теоретиков, как и многие другие запреты в химии, был снят, поскольку экспериментаторами были получены соединения непереходных элементов не только с одинарными, но двойными и даже с тройными связями. Ими стали металлорганические соединения 12 группы с одинарными связями М-М (например, [Zn+1R]2 , рис. 4 б) и, конечно, многочисленные соединения 13 группы, получаемые практически одним методом - восстановлением металлорганических производных MR3 с стерически затрудненным R щелочными металлами, и гетерометаллические соединения 12 и 13 групп. Структура одного из них – гетероядерного комплекса цинка и галлия с одинарной связью М-М приведена ниже.
Из соединений с двойными связями получен, например, комплекс диборена L(H)B=B(H)L. К настоящему времени вершиной этих работ являются ионные комплексы состава Na2[RMMR], где M-Al, Ga, в которых постулируется тройная связь М-М. Так, в галлиевом комплексе расстояние М-М равно 2.32 Å при сумме ковалентных радиусов 2.52 Å. Это кратчайшее из известных расстояний Ga-Ga.. Но квантово-химического обоснования этих фактов, прежде всего объяснения причин и вообще объяснения факта и возможности существования тройной связи у непереходных металлов, пока не имеется.
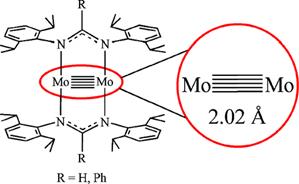
Рис. 4. а) «Пятерная» связь Мо-Мо в биядерном комплексе
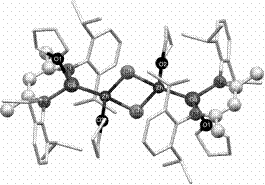
б) Одинарная связь Zn-Zn в биядерном комплексе
Донорно-акцепторная связь p и s-типов, образуемая при донировании электронной плотности с молекулы донора на вакантные орбитали кислого (по Льюису) центра (обычно атом или ион металла, но могут быть и другие варианты). В курсах химии она обычно рассматривается в разных разделах теории химической связи: как ковалентная связь, как самостоятельная связь и как связь, описывающая один из типов межмолекулярного взаимодействия (ниже). Подобная трактовка ее природы обусловлена попытками выделить «оригинальное» начало в ее возникновении. Однако следует иметь ввиду, что в большинстве случаев такое «объяснение» приводит к результату не отличимому от описания полярной ковалентной связи. Например, в тетраэдрических катионе NH4+, анионе AlH4- или нейтральной молекуле ZnS невозможно различить, какая связь исходно ковалентная, а какая по своей природе д.а. или по терминологии Вернера “дополнительная”, поскольку все связи N-H, Al-H и Zn-S относятся к s-типу, все расстояния и энергии связей в этих ионах одинаковы, одинаковы и схемы МО, описывающие их образование, хотя в целом все они несколько длиннее истинно ковалентных и, соответственно, слабее. Эта неразличимость двух типов связей, лежащая в основе описания химической связи в комплексных соединениях (метод валентных связей), более подробно рассматривается ниже.
Дативная связь возникает как дополнительная или донорно-акцепторная к ковалентной в ряде металлорганических и комплексных соединений. Ее можно описать как простую, возникающую при дополнительном (помимо “основной” д.-а. связи) донировании электронных пар (пары) от лиганда на вакантные орбитали центрального атома, например в комплексах с лигандами OH-, OR-, NR2- и т.п., в которых атомы О и N имеют по две пары несвязанных электронов, так и обратную, поскольку в ней донирование электронов идет от металлического центра на вакантные p-орбитали лиганда, например от атома переходного металла к молекуле СО в некоторых карбонилах. Возникающая при этом двоесвязанность или повышение кратности связи (хотя этот факт не следует рассматривать и описывать в терминах двойной связи), приводит к заметному укорочению межатомных расстояний металл-лиганд, т.е. к усилению межатомного взаимодействия и увеличению прочности связи по сравнению с взаимодействием в соединениях с чисто ковалентным связыванием.
Агостическая связь – один из типов внутримолекулярной связи, возникающей при взаимодействии электронной плотности на удаленном атоме молекулы с вакантной орбиталью на другом атоме, обычно на центральном атоме металла. На рис. 5 приведен пример образования агостической связи в ходе реакции восстановления атома Ti(IV) до Ti(III)и образования биядерного гидридного комплекса, в котором имеются две «нормальные» трехцентровые связи Ti-H-B и одна мостиковая связь с удлиненными расстояниями С-Н и Н-Ti.
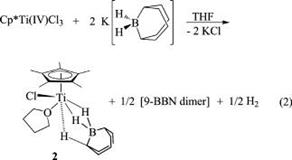
Рис.5. Образование агостической связи C-H-Ti в биядерном борогидридном комплексе титаноцена.
Наиболее типично образование агостической связи в металлорганических соединениях с длинными алкильными радикалами с избыточной плотностью на концевых протонах. Сильное агостическое взаимодействие может сопровождаться реакцией окислительного присоединения, приводящей к повышению степени окисления атома металла на две единицы и к образованию гидрида.
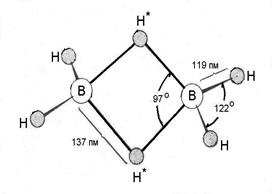
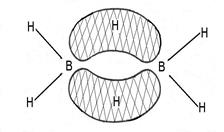
Самыми необычными в настоящий момент остаются многоцентровые ковалентные связи (иногда говорят координационные связи), образованные не в результате спаривания электронов от двух атомов, а в результате обобществления двух электронов тремя или более центрами, т.е. образование многоцентровых двухэлектронных связей. Таким образом, изначально с точки зрения теории Льюиса эта связь, будучи электронодефицитной, не могла существовать, поэтому вопросы строения ряда соединений, таких как бороводороды и первого представителя этого ряда диборана (рис. 6), металлорганические соединения, галогениды и гидриды легких металлов, катиона
а)
б)
Рис. 6. Структура В2Н6 по данным электронографии (а) и в графическом исполнении как представителя структур с «банановым» типом связи (б).
метония –СН5 и большом числе других органических и неорганических соединениях долгое время оставались загадочными и были предметами острейших дискуссий. В настоящее время описание многоцентровых связей базируется на методе МО (рис. 7 и 8). Например, при взаимодействии двух атомов бора и атома водорода каждый из них
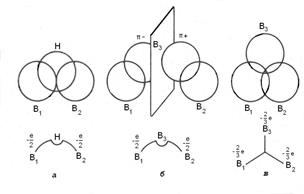
Рис. 7. Три типа трехцентровых связей возможных в бороводородах: а-открытая В-Н-В связь; б- открытая В-В-В связь; в- центральная В-В-В связь.
представляет по одной орбитали, в результате чего возникают две разрыхляющие и одна связывающая орбиталь, которая и заполняется парой электронов, представляемых атомом водорода (1 электрон) и двумя атомами бора (условно по 0.5 электрона от каждого).
При образовании трехцентровой связи атом бора формально приобретает законченную оболочку ближайшего инертного газа. Естественно, что каждый фрагмент В-Н в подобной или, как ее иногда называют, “банановой” связи В-Н-В, в которой пара электронов делокализована между тремя центрами, менее прочен, чем “классическая” двухцентровая локализованная связь В-Н (это видно из расстояний B-H в мостиковых и концевых положениях рис. 6). Но в целом, возникающая при этом молекула диборана,
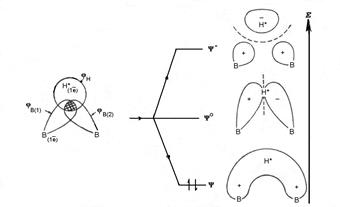
Рис. 8. Образование трехцентровой двухэлектронной связи В-Н*-В и энергетическая диаграмма молекулярных орбиталей.
вполне устойчива и именно она, а не мономерный боран существует как индивидуальное вещество.
Другой яркий пример стабилизации молекулы, происходящий за счет образования многоцентровой координационной связи, образование при взаимодействии хлористого железа с циклопентадиенидом натрия не s, а p-комплекса, т.н. ферроцена – удивительно стабильного соединения, в котором атом железа связан с 10 атомами углерода.
Металлическая связь
И, наконец, третий самостоятельный тип химической связи - металлическая связь, образующаяся в результате взаимодействия электронного газа, состоящего из валентных электронов, с остовом кристаллической решетки, состоящем из положительно заряженных ионов металла, находящихся в динамическом равновесии с незаряженными атомами. Другое, несколько неожиданное определение металлической связи – это ковалентная ненасыщаемая и ненаправленная связь. Таким образом, в металле каждый электрон как бы принадлежит всем атомам одновременно, а кристалл металла можно рассматривать как одну гигантскую молекулу, в которой все атомные орбитали превращаются в молекулярные орбитали, распределенные или делокализованные по всей молекуле и при этом незначительно различающиеся по энергиям. Это является следствием того, что в металлах и подобных им соединениях запрещенная зона, разделяющая валентную зону и зону проводимости, исчезающе мала, поскольку для этих веществ она составляет сотые доли eV, тогда как для изоляторов или ковалентно связанных веществ превышает 5 eV. В результате электронный газ свободно переходит из соприкасающихся или даже смешанных зон практически без затрат энергии. Отсюда следует, что в металлах полностью отсутствует ковалентная составляющая, чего нет даже в ионных соединениях.
Электропроводность в металлах с повышением температуры, хотя и слабо, но уменьшается, а проводимость полупроводников и диэлектриков резко возрастает. Это различие определяется различной концентрацией носителей заряда n в уравнении σ=neμ (σ- электропроводность в Ом-1·см-1, n- концентрация носителей заряда, е- величина заряда, μ – подвижность носителей заряда), которая не меняется с температурой в металлах, но быстро растет в диэлектриках и полупроводниках, тогда как μ во всех случаях слабо уменьшается с температурой. Таким образом, в полупроводниках и диэлектриках рост n намного опережает падение μ.
Электронную структуру металлов и многих других твердых тел обычно описывают в рамках зонной теории. В этой теории все валентные электроны металлов занимают энергетические уровни, которые делокализованы по всей кристаллической решетке и каждый из валентных подуровней (s, p и d) как бы образуют одну гигантскую молекулярную орбиталь или s-валентную зону, p-валентную зону или d-валентную зону. В рамках этих представлений металлы, полупроводники и диэлектрики различаются по (i) зонной структуре, (ii) по степени заполненности валентных зон и (iii) по ширине запрещенной зоны, расположенной между заполненной и пустой зонами.
В “химическом” подходе (теория «плотной химической связи») зонная теория описывается в терминах теории молекулярных орбиталей, рассматривающей небольшие молекулы конечных размеров, но распространенной на случай бесконечных трехмерных структур (рис. 9). Подобный подход ведет к неограниченному увеличению количества МО (в металлах ~6·1023) и, как следствие, к уменьшению энергетических различий между
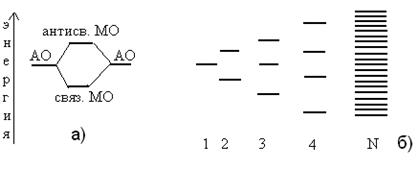
 Рис. 9. Молекулярные орбитали в двухатомной молекуле (а) и расщепление энергетических уровней согласно теории молекулярных орбиталей (б)
Рис. 9. Молекулярные орбитали в двухатомной молекуле (а) и расщепление энергетических уровней согласно теории молекулярных орбиталей (б)
соседними орбиталями и энергетического зазора между связывающей и антисвязывающей орбиталями. В этих условиях нет смысла рассматривать каждый из энергетических уровней как одну МО, поэтому говорят об энергетических состояниях.
В качестве примера рассмотрим схему зонной структуры металлического натрия, рассчитанную из теории «плотной химической связи» (рис. 10). Видно, что ширина каждой
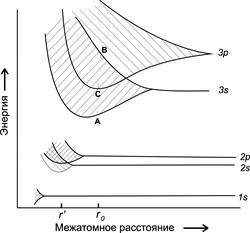
Рис. 10. Схема зонной структуры металлического натрия
отдельной энергетической зоны зависит от расстояния между атомами, которое определяет и степень перекрывания орбиталей соседних атомов. При межатомном расстоянии rо (расстояние между атомами в решетке металлического натрия) верхние уровни 3s-зоны и нижние уровни 3p-зоны соседних атомов существенно перекрываются, образуя широкие (3s+3p)-зоны, между которыми отсутствует энергетическая щель. При межатомном расстоянии ro 1s, 2s и 2p-орбитали соседних атомов не перекрываются, т.е. не образуют зоны, сохраняя дискретные атомные орбитали. Если каким-либо способом удалось бы сблизить атомы до расстояния r1<rо, то 2s и 2p орбитали перекрылись бы, образуя (2s+2p) энергетические зоны, но при этом 1s-орбитали оставались бы атомными.
Возникают вопросы: можно ли это сделать, и, если можно, то, как и, вообще, может ли одно и тоже вещество, находясь в одном и том же фазовом состоянии, менять тип химической связи? При обычных земных условиях это, конечно, осуществить невозможно. В лучшем случае, например, при нагревании, мы будем наблюдать фазовый переход (без изменения агрегатного состояния), в результате которого может произойти некоторое уменьшение или увеличение доли одной из составляющих химической связи, например, увеличится ионная составляющая, но в целом никакого радикального изменения в способе связывания не произойдет. Иное дело, когда будет происходить изменение другого термодинамического параметра – давления. Причем это изменение может осуществляться одновременно с изменением температуры или протекать в изотермических условиях.
Сжимаемость вещества зависит от различных причин, начиная от свойств индивидуальных атомов и составленных из них молекул и кончая типом связи в этом соединении. В первом приближении малой сжимаемостью и соответственно малыми степенями сжатия, а отсюда и незначительными объемными эффектами обладают вещества, имеющие достаточно большую свободную энергию образования и «жесткую» кристаллическую структуру. Это, например, касается соединений с ионным типом связи - оксидов, фторидов и хлоридов металлов. Так, ионные галогениды щелочных металлов не изменяют типа связи даже при их сжатии до сотен Кбар и сохраняют при этом все свои физические свойства. Их барический полиморфизм характеризуется величинами объемных эффектов не более 10 % и только при значениях DP> 1000 Кбар (100 GPa), при которых уже можно говорить об изменении электронной структуры вещества, иногда связанном с очень глубоким изменением конфигурации внешних валентных электронных уровней вплоть до их инверсии. Как правило, подобные изменения приводят к металлизации вещества. Подобная трансформация свойств была предсказана в начале XX века Д. Гольдхаммером, К. Херцфельдом и Н. Моттом и затем блестяще подтверждена экспериментальными работами ученых, в основном физиков, многих стран, в том числе и работами нашего соотечественника Леонида Федоровича Верещагина – основателя кафедры физики и химии высоких давлений. Если ионные соли требуют для этого превращения очень высокие давления, то многие металлиды, такие как фосфор, мышьяк, селен, и другие уже при 1-10 ГПа образуют зоны и приобретают свойства металлов, т.е. изменяют тип связи на металлический. Так, белый фосфор, построенный из тетраэдрических молекул Р4, при 3,5 ГПа превращается в слоистую модификацию черного цвета, которая в свою очередь при 10 ГПа переходит в другую еще более плотную слоистую модификацию с контактом Р-Р=2.9 Å (в исходном белом это расстояние равно 3.87 Å). При 12-13 ГПа образуется изотропная кубическая модификация с расстоянием Р-Р=2.38 Å и окатаэдрическим окружением, которая при повышении давления до 15-20 ГПа переходит в вещество с металлическим типом проводимости.
Другой пример – иод, гантелеобразные молекулы которого при 20 ГПа образуют искаженную слоистую структуру, симметризующуюся при 55 ГПа в плотноупакованную изотропную модификацию со всеми свойствами металла. При этом его плотность возрастает в 2 раза по сравнению с обычной модификацией. Возможно, что аналогичный эффект, вызванный перекрыванием 1s-орбиталей и образованием 1s-зон, т.е. переходом в металлическое состояние, может иметь место и при сдавливании водорода. Впервые на такую возможность указали физики-теоретики в 1935 г. E.Wigner и H.B. Huntington, посчитав, что для этого будет достаточно давления в 25 ГПа. Но уже в 60 годах стало очевидным, что они ошиблись, как минимум, на порядок и сейчас считается, что для этого превращения требуются давления более 300 ГПа, которые, например, существуют близко к ядру планеты-гиганта Юпитера. Рекорд земных ученых 342 ГПа при 300К на алмазных наковальнях. Но даже при этих условиях воспроизводимых результатов по металлизации водорода не наблюдали и не получено подтверждений ранним результатам по синтезу металлического водорода в СССР и в США в 80-90 г.г. Следует отметить, что существует и другая гипотеза, основанная на численных метода, согласно которым водород при всех давлениях и всех температурах существует в виде жидкости, но жидкости квантовой и к тому же сверхпроводящей. При этом он остается в молекулярной форме, хотя ранее предполагалось, что при переходе в металлическое состояние водород атомизируется. Считается, что подобная жидкость являет собой принципиально новое состояние материи, однако эта точка зрения еще не нашла независимого экспериментального подтверждения.
Таким образом, на сегодняшний день возможность металлизации чистого водорода остается весьма призрачной. Но для решения этой задачи экспериментаторы пробуют иные подходы, связанные, например, с использованием в процессах металлизации соединений с высокой п
Дата добавления: 2016-07-18; просмотров: 3114;
Поиск по сайту
Узнать еще
- II степень (сегментарная дистопия матки)
- II. Степень насыщенности, определяемая природой связи между атомами углерода.
- III степень (тотальная дистоция матки)
- Автоматическая локомотивная сигнализация непрерывного действия с числовым кодированием (АЛСН)
- Бензиновый двигатель внутреннего сгорания со сверхвысокой степенью сжатия.
- Важнейшие классы числовых функций одной переменной.
- Валентность и степень окисления

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
