Клинический пример 9
Больной 68 лет. Находился на санаторном лечении. Состояние после аортокоронарного шунтирования (АКШ). Принимает варфарин.
Коагулограмма для контроля антикоагулянт-ной терапии: количество тромбоцитов 330 х 109/л.
В моче микрогематурия (2-3 эритроцита в поле зрения).
ПВ 80,5с (норма 15,5-21,2 с), MHO8,5.
Заключение: передозировка варфарина. Необходимо уменьшить дозу. Контроль MHO проводить ежедневно до достижения рекомендуемого для профилактики тромбоэмболии значения MHO = 2-3.
Патология гемостаза
Гепариноподобные антикоагулянты
Развитие гепариноподобного антикоагулянта было описано у пациентов, страдающих неопла-зиями или получающих терапию сурамином при адренокарциноме. Клинически это нарушение проявляется выраженными геморрагическими симптомами. Лабораторно выявляется удлинение тром-бинового времени, которое корригируется применением протаминсульфата, либо толуидинового голубого, либо гепариназы. Биохимические и физико-химические исследования показали сходство этого антикоагулянта с гликозаминогликанами.
Заболевания печени
Кровотечения часто сопутствуют хроническим или острым заболеваниям печени. Геморрагичес-
кий синдром при заболеваниях печени носит смешанный характер (по гематомному и микроцир-куляторному типу). Наиболее опасны кровотечения из пищеварительного тракта, которые нередко становятся причиной гибели этих пациентов.
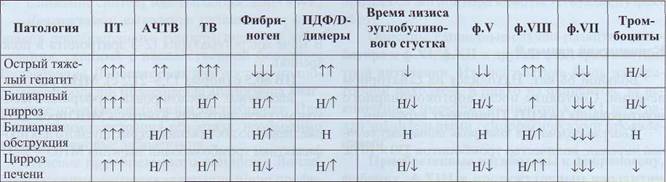
В табл. 60 перечислены основные дефекты системы гемостаза, связанные с поражением печени.
В табл. 61 представлены особенности результатов лабораторного анализа гемостаза при различных заболеваниях печени.
Системный фибринолиз
Значительная активизация системного фиб-ринолиза - довольно редкая причина геморрагического синдрома. Однако развитие системного гиперфибринолиза может повлечь за собой
Дефекты системы гемостаза при поражениях печени
Таблица 60

|
Возможные изменения показателей гемостаза при заболеваниях печени
Таблица 61

|
Патология гемостаза
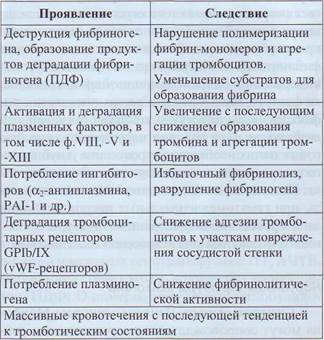
опасные геморрагические проявления, а при истощении плазминогена - тромбозы (табл. 62). Системный фибринолиз может быть причиной массивных кровотечений из желудочно-кишечного тракта у пациентов с печеночной недостаточностью.
Таблица 62
Гиперфибринолиз и его осложнения
Большинство скрининговых тестов не выявляет состояние гиперфибринолиза. Относительно специфичными являются тесты определения времени лизиса сгустка. Тромбоэластограмма способна наглядно продемонстрировать развитие гиперфибринолиза.
Из-за относительно невысокой специфичности плазмин может деградировать многие белки крови. Кроме того, плазмин может активировать металлопротеазы, которые, в свою очередь, способны индуцировать деструкцию тканей и апоп-тоз. Высока вероятность развития гиперфибринолиза при множественных травмах, сепсисе, ДВС-синдроме, выпадении функции органов, обширном метастазировании с деструкцией тканей. Врожденный или приобретенный недостаток одного или нескольких ингибиторов фибринолиза (особенно α2-антиплазмина или PAI-1) сопровождается проявлениями гиперфибринолиза.
Гиперфибринолиз клинически проявляется склонностью к кровотечениям, а при истощении факторов - тромбозами. Состояние гиперфибринолиза необходимо диагностировать и лечить. Для коррекции гиперфибринолиза используются апротинин, ингибиторы фибринолиза и ингибиторы протеолитических ферментов.
Геморрагические мезенхимальные лисплазии
Мезенхимальные дисплазии (МД) - группа врожденных заболеваний соединительной ткани, в основе которых лежит недостаточное или аномальное развитие коллагеновых структур, приводящее к неполноценности сосудистой стенки, связочного аппарата, клапанов сердца, кожи, скелета и других стромальных образований, часто сочетающихся с неполноценностью иммунитета и гемостаза.
Мезенхимальные дисплазии, сочетающиеся с нарушениями в системе гемостаза и геморрагическим синдромом, в современной литературе обозначают как геморрагические мезенхимальные дисплазии (ГМД). Геморрагические проявления описаны при многих мезенхималь-ных дисплазиях: генерализованной фибродисп-лазии (синдром Черногубова-Элерса-Данлоса), мезодермальной аномалии Марфана, несовершенном остеогенезе, синдроме отсутствия лучевой кости (ТАР-синдроме), мозжечковой атак-сии-телеангиэктазии (синдром Луи-Бар), болезни Рэндю-Ослера, диффузной ангиокератоме туловища (болезнь Фабри), гемангиомах (синдром Казабаха-Меритта, микроангиоматозы с тром-боцитопенией) и др. В основе геморрагического синдрома при гематомезенхимальных дисплазиях лежат несколько механизмов:
• Нарушение строения соединительной тка
ни приводит к повышенной ранимости со
судов.
• Сочетание генетически обусловленных ано
малий коллагена с генетически обусловлен
ными нарушениями компонентов системы
гемостаза (качественные и/или количествен
ные дефекты тромбоцитов, дефицит актив
ности факторов свертывания, качественные
и/или количественные дефекты фактора
Виллебранда, нарушения взаимодействия
Патология гемостаза
 | |||||
 | |||||
 | |||||
тромбоцитов с коллагеном сосудов, качественные и/или количественные дефекты фибриногена, в том числе нарушение его полимеризации).
• Сочетание аномалии строения коллагена с приобретенными нарушениями системы гемостаза (секвестрация тромбоцитов и потребление факторов свертывания в геман-гиомах, вторичные дефекты системы гемостаза на фоне хронических инфекций у пациентов с иммунодефицитными состояниями, в частности при синдроме Луи-Бар). Несколько подробнее остановимся на двух состояниях.
Гемангиомы,особенно гигантскиекавернозные гемангиомы Казабаха-Меритта, приводят к активации и секвестрации тромбоцитов, потреблению плазменных факторов гемостаза. Помимо кровотечений вследствие травмы патологически измененных сосудов, изменения гемостаза вносят свой вклад в развитие геморрагического синдрома у этих пациентов.
Одним из серьезных осложнений больших гемангиом является острый или хронический ДВС, приводящий к потреблению прокоагулян-тов и активации фибринолиза. При этих явлениях нарушения гемостаза характеризуются тромбоцитопенией, снижением концентрации фибриногена, повышением количества ПДФ, D-димеров. На фоне этого часто развивается анемия.
Болезнь Рэндю-Ослера-Вебера - наследственное заболевание, проявляющееся множественными телеангиэктазиями на коже, в желудочно-кишечном тракте, дыхательных путях и других органах. Области телеангиэктазий легко ранимы, вследствие чего у пациентов возникают носовые и желудочно-кишечные кровотечения.
Причиной заболевания является мутация расположенного в 9-й хромосоме гена эндоглина -белка, участвующего в ангиогенезе и репарации тканей. Распространенность заболевания составляет 1:2500-40 000 человек.
Специфических изменений при исследовании свертывающей системы крови нет, однако болезнь Рэндю-Ослера-Вебера может сочетаться с болезнью Виллебранда.
Диагностика гематомезенхимальных диспла-зий основана на сочетании тщательного клини-
ческого обследования пациента и комплекса лабораторных методов.
Спектр лабораторных исследований, применяемых для уточнения характера патологии гемостаза, очень широк. Помимо обязательного проведения скрининговых тестов, в диагностическую палитру необходимо включить исследование функции тромбоцитов (агрегация с различными индукторами, исследование адгезии тромбоцитов, определение доступности фактора 3 тромбоцитов, тест ретракции кровяного сгустка), при исследовании плазменных белков системы свертывания крови обязательно исследование концентрации фибриногена по Клауссу, анализ процесса аутопо-лимеризации фибриновых мономеров, активности фактора Виллебранда в тестах ристоцетин-ко-факторной активности и коллаген-связывающей активности. При наличии нарушений в скрининговых тестах необходимо проведение углубленного исследования соответствующего звена гемостаза. Помимо анализа состояния системы гемостаза, при гематомезенхимальных дисплазиях показано проведение исследования структуры коллагена и молекулярно-генетическое исследование.
Нарушения структуры коллагена
Синдром Элерса-Данлоса, синдром Марфа-на могут сопровождаться геморрагическим синдромом различной тяжести. Лабораторные нарушения при этой патологии могут отсутствовать. Однако может быть удлинение времени кровотечения, умеренное нарушение функции тромбоцитов, признаки дисфибриногенемии.
Кровотечения, связанные с массивной кровопотерей
Быстрая массивная кровопотеря - нередкое осложнение тяжелых травм и патологических родов; она может осложнять операции трансплантации органов, оперативное лечение сердечно-сосудистой патологии и рака. Это состояние может требовать массивных гемотрансфузий (более чем 1 ОЦК в сутки). При кровопотере интенсивностью 1 ОЦК менее чем за 2 часа могут возникать серьезные нарушения гемостаза.
Имеется несколько патогенетических факторов этих нарушений.
Патология гемостаза
Наиболее распространенная клиническая проблема - разведение плазменных компонентов гемостаза и тромбоцитов вводимыми плазмоза-менителями. Использование коллоидных и крис-таллоидных растворов, эритроцитарной массы, разведенной изотоническим солевым раствором В гематокрита 60%, при массивной гемотранс-фузии приводит к значительному снижению активности компонентов гемостаза.
Одним из следствий массивной кровопоте-ри и массивной заместительной терапии являются тромбоцитопения и тромбоцитопатия, возникающие вследствие быстрой внутрисосу-дистой активации тромбоцитов продуктами деградации фибрина/фибриногена, разведения и секвестрации тромбоцитов в сосудистом русле на фибриновых депозитах. В случаях массивной кровопотери при тяжелой травме, особенно головы и мозга, гипотонии с гипоксией и ацидозом, бактериальном сепсисе, преждевременной отслойке плаценты может развиться ДВС-синдром.
Рекомендуемые лабораторные тесты для контроля состояния гемостаза при массивной крово-потере и массивных гемотрансфузиях: ПТ, АЧТВ, фибриноген, гематокрит, количество тромбоцитов, ПДФ, D-димеры, тест лизиса эуглобулино-вого сгустка.
Нарушения гемостаза, связанные с патологией почек
До начала использования гемодиализа кровотечения были серьезным осложнением у пациентов с хроническим поражением почек. Однако даже при систематическом проведении гемодиализа примерно у половины больных с хронической почечной недостаточностью имеются такие проявления, как пурпура, меноррагии, носовые кровотечения, реже кровотечения из желудочно-кишечного тракта. Тяжелые кровотечения у данной группы пациентов, как правило, связаны с травмой или оперативным лечением, тем не менее геморрагический синдром осложняет их ведение и в других ситуациях.
Патофизиологические механизмы, приводящие к геморрагиям у пациентов с хронической почечной недостаточностью, в основном связаны с явлениями уремии. У этих пациентов
выявляются значительные нарушения тромбо-цитарно-сосудистого взаимодействия. Одним из возможных механизмов является повышение синтеза и экспрессии оксида азота эндотелием. Определенный вклад вносит анемия; патогенетические механизмы этого неясны, однако после трансфузии эритроцитарной массы уменьшается время кровотечения и геморрагический синдром.
Еще одним патогенетическим фактором повышенной кровоточивости при хронической почечной недостаточности является тромбоцитопения, которая довольно часто бывает у таких пациентов.
Процедура гемодиализа сопровождается применением гепарина. Остаточное его количество может вносить вклад в геморрагические проявления.
Лабораторные данные
У пациентов с уремией часто удлинено время кровотечения. Степень удлинения может варьировать от незначительной до очень большой. Агрегация тромбоцитов может быть снижена, однако ее снижение не коррелирует с тяжестью геморрагических проявлений. Другие скрининго-вые тесты (ПТ, АЧТВ, тромбиновое время, фибриноген) могут быть нормальными или немного удлинены.
У пациентов с нефротическим синдромом, особенно у детей, изменение скрининговых тестов возникает даже без уремии за счет потери ф.IХ и ф.ХII через почки.
Тромботические проявления у пациентов с поражением почек ассоциируются с нефротическим синдромом, гипергомоцистеинемией и гепа-рин-индуцированной тромбоцитопенией.
Амилоидоз
Около 10% пациентов с системным амилои-дозом имеют выраженный геморрагический синдром. Безусловно, кожный гемосиндром у них может быть связан с повышением хрупкости сосудов в связи с отложением амилоида в их стенках и в периваскулярном пространстве. Кроме того, у этих больных имеются также системные дефекты гемостаза, в первую очередь - снижение активности ф.Х. Вероятной причиной этого является адсорбция ф.Х на амилоиде. Активность
Патология гемостаза
ф.Х в плазме пациентов с амилоидозом может составлять 2-4% от нормальных значений.
При данном заболевании также отмечается усиление системного фибринолиза. Полностью механизм этого процесса не раскрыт. Происходит снижение оц-антиплазмина, видимо, тоже за счет адсорбции его на амилоиде. Описано повышение в плазме активаторов плазминогена и сни-
жение ингибитора активатора плазминогена-1. Наконец, имеются единичные описания развития специфического ингибитора к ф.VIII.
Лабораторная диагностика включает стандартные тесты скрининга (ПТ, АЧТВ, ТВ, фибриноген, время кровотечения, количество тромбоцитов), помимо этого, рекомендуется проводить исследование рептилазного времени.
Тромботические заболевания
Тромбоцитоз
Увеличение содержания тромбоцитов в сыворотке свыше 400 х 109/л определяется как тромбоцитоз. Тромбоцитоз возникает при миелопро-лиферативных заболеваниях, злокачественных состояниях (рак, болезнь Ходжкина, лимфомы), воспалениях (ревматоидный артрит, язвенный колит, туберкулез, остеомиелит), после удаления селезенки (2 месяца) и при другой патологии. Тромбоцитоз - компонент острофазной реакции. Эссенциальная тромбоцитемия - миелопролифе-ративное заболевание. Состояние после спленэк-томии объясняется удалением основного места разрушения тромбоцитов. Увеличенное содержание тромбоцитов может привести к артериальному или венозному тромбозу.
Тромбофилии. Общее представление
Проблема патологического тромбообразова-ния - одна из важнейших терапевтических проблем в развитых странах. Ишемическая болезнь сердца, тромбоэмболия легочной артерии (ТЭЛА), тромбозы глубоких вен - патологические состояния, приводящие к тяжелой инвалидности и гибели человека. При лечении таких пациентов лабораторный контроль состояния гемостаза- важнейший фактор успеха.
Артериальные и внутрисердечные тромбы, образуясь в условиях высокой скорости кровотока, состоят преимущественно из тромбоцитов, соединенных фибриновыми мостиками - белые тромбы. Артериальные тромбы преимущественно пристеночные. Важнейшими факторами патогенеза артериального тромба являются врожденная или приобретенная аномалия сосудистой
стенки и патологическая активация тромбоцитов. Наиболее частая аномалия - атеросклероз. Другие состояния - это врожденные нарушения развития сосудов, ангиоматозные образования, инфекционное поражение эндотелия, ятрогенные нарушения. Закупорка просвета артериального сосуда может наступить при увеличении тромба на фоне нарастания патологического процесса (атеросклероз) или при эмболии нижележащих более мелких сосудов оторвавшимися элементами тромба.
Венозные тромбы образуются в условиях относительно медленного кровотока и низкого напряжения сдвига, включают в себя значительное количество эритроцитов и большое количество фибрина. Венозные тромбы часто полностью об-турируют просвет сосуда. Основной механизм образования венозного тромба связан с повышением свертываемости крови и стазом. Если тромбоз возникает в магистральной вене, нарушение венозного оттока может привести к венозному полнокровию, повышению внутриорганного давления, снижению притока крови, ишемии и дистрофическим изменениям органа. При сохраняющемся кровотоке по тромбированной вене возможен отрыв части тромба и эмболия.
Патогенез тромбофилии
Как правило, тромбофилия - комбинированное состояние, возникающее вследствие действия нескольких патогенетических факторов. Основные патогенетические факторы тромбофилии: • Повреждение эндотелиальных клеток с обнажением тромбогенных субэндотелиальных структур.
Патология гемостаза
• Активация тромбоцитов циркулирующими
агонистами либо вследствие взаимодействия
тромбоцитов с субэндотелиальными структу
рами или фактором Виллебранда.
• Активация свертывания крови.
• Резистентность к антикоагулянтам или дефи
цит антикоагулянтов.
• Снижение активности фибринолиза.
• Реологические нарушения и стаз.
Эти факторы возникают на фоне целого ряда патологических состояний.
Наследственные факторы риска патологического тромбообразования (генетические дефекты выявляются у 30-50% пациентов с тромботичес-ким состоянием):
• Мутация фактора V (фактор V Лейден).
• Дефицит антитромбина III.
• Дефицит протеина С.
• Дефицит протеина S.
• Мутации протромбина, в первую очередь
G20210A.
• Полиморфизм тромбоцитарного рецептора
GPIIIa.
• Дисфибриногенемии.
• Гиперлипопротеинемия (а).
• Мутация ингибитора пути тканевого факто
ра (ИВП), 536С/Т.
• Гипергомоцистеинемия (у детей, как прави
ло, носит наследственный характер).
• Дефекты тромбомодулина.
• Мутации гена GPIIIa тромбоцитов.
• PAI-1.
Приобретенные факторы патологического тромбообразования:
• Возраст.
• Пороки сердца и сосудов.
• Атеросклероз.
• Катетеризация вен, особенно длительное на
хождение катетера в вене.
• Повышение вязкости крови (полицитемия,
потеря жидкости).
• Операция или травма.
• Длительная иммобилизация.
• Инфекция (ВИЧ, ветряная оспа, гнойный
тромбофлебит).
• Аутоиммунные заболевания (волчаночный
антикоагулянт, антифосфолипидный синд
ром, сахарный диабет, болезнь Бехчета и др.).
• Нефротический синдром.
• Ингибиторы к протеинам S и С.
• Онкологические заболевания.
• Химиотерапия (L-аспарагиназа, преднизо-
лон).
• Заболевания печени.
• Талассемия (постспленэктомический тромбоз
печеночных вен).
• Серповидно-клеточная анемия.
• Прием гормональных противозачаточных
препаратов.
Факторы, роль которых в развитии тромбозов неясна:
• Высокий уровень активности факторов VIII,
XI, XII, Виллебранда, ингибитора активато
ра плазминогена.
• Дефицит факторов XII, кофактора гепари
на II, плазминогена, активаторов плазмино
гена, тромбомодулина.
Дата добавления: 2016-08-06; просмотров: 2417;
Поиск по сайту
Узнать еще

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
