Порядок выполнения работы
Вариант 1. Определение удельной поверхности адсорбентов по удерживаемому объему.
Для проведения работы необходимы:
Ø хроматограф;
Ø микрошприц;
Ø адсорбент, например силикагель;
Ø адсорбат, например бензол или гептан.
Для выполнения работы может быть использован любой газовый хроматограф с катарометром в качестве детектора. Удобнее работать с хроматографом, в котором проба вводится в одну из двух равноценных колонок, а вторая колонка является сравнительной (подается чистый газ-носитель). Для того чтобы ячейки детектора работали в идентичных условиях, т. е. чтобы электрическое сопротивление элементов детектора определялось только составом газовых потоков, скорость газа-носителя через них должна быть примерно одинаковой (допустимое отклонение ±2 мл/мин).
Одну из колонок хроматографа (колонка 1) заполняют силикагелем (масса m1) с известной удельной поверхностью Sуд1, а вторую – адсорбентом (масса m2), удельную поверхность которого необходимо определить. Условия хроматографического анализа: длина колонки 1 м, внутренний диаметр 3 мм; температура термостата колонок 160 или 180 °С для бензола или гептана соответственно; газ-носитель – азот, его расход 30 мл/мин; скорость движения диаграммной ленты КСП – 12 мм/мин.
Работу выполняют в следующей последовательности. Включают хроматограф (под руководством преподавателя) и регулируют скорость газа-носителя v в хроматографических колонках. Затем с помощью микрошприца в хроматографическую колонку 1 вводят 5–7 раз по 2 мкл адсорбата (момент ввода пробы отмечается на диаграммной ленте. Каждую последующую пробу вводят только после полного проявления пика, соответствующего предыдущей пробе. Аналогичным образом вводят адсорбат в колонку 2 с адсорбентом, удельную поверхность которого определяют.
Рассчитывают значения tr как отношения расстояний l, пройденных пером самописца от момента ввода пробы (точка О на рис. 3.3) до середины пиков (точка А), к скорости движения диаграммной ленты самописца. Затем по уравнениям (3.25) – (3.28) рассчитывают величины t0, V¢r и Vm. По уравнению (3.31) вычисляют удельную поверхность адсорбента в колонке 2. Для расчета используют средние значения Vm,1 и Vm,2, найденные по результатам нескольких измерений. Полученные данные записывают в таблицу (см. табл. 3.3).
Таблица 3.3 – Экспериментальные и расчетные данные по определению удельной поверхности адсорбента
| Номер колонки | U, мл/мин | l, мм | tr, мин | Vr, мл | V¢r, мл | m, мм | Vm, мл/г | Sуд, м2/г |
Вариант 2. Определение удельной поверхности адсорбентов методом тепловой десорбции азота.
Для проведения работы необходимы:
Ø хроматографическая установка;
Ø секундомер;
Ø адсорбент;
Ø азот, гелий.
Блок-схема хроматографической установки, используемой для определения удельной поверхности адсорбентов методом тепловой десорбции, представлена на рис. 3.4.
Потоки гелия и азота из баллонов 1 и 2 подаются в определенном соотношении в смеситель 3, из которого газовая смесь поступает в сравнительную камеру детектора 6 и далее в колонку 8 с исследуемым адсорбентом, в которой при охлаждении происходит адсорбция азота. Из колонки газовая смесь поступает в измерительную камеру детектора 7. Детектор фиксирует изменение состава газовой смеси в результате адсорбции. Сигнал детектора поступает на самопишущий потенциометр 5.
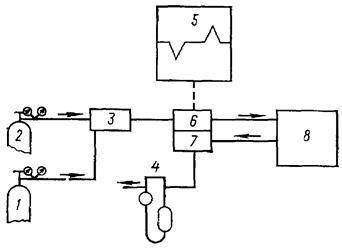
рисунок 3.4 – Блок-схема хроматографической установки для определения удельной поверхности методом тепловой десорбции:
1,2 – баллоны с газами; 3 – смеситель; 4 – пенный расходомер;
5 – потенциометр; 6,7 – камеры детектора; 8 – колонка.
Объемная скорость газовой смеси должна быть постоянной при всех значениях объемной доли азота в смеси. Скорость подачи смеси контролируют на выходе из детектора пенным расходомером 4.
Условия хроматографирования: длина колонки – 0,1 м; диаметр колонки – 3 мм; скорость газового потока через колонку – 30 мл/мин; начальное содержание азота в газовой смеси составляет 5 % (об.).
Включают хроматограф и с помощью редукторов на баллонах 1 и 2 устанавливают необходимую скорость подачи газовой смеси. При отсутствии охлаждения хроматографической колонки через сравнительную и измерительную камеры детектора проходит газовая смесь одинакового состава, поэтому вначале перо самопишущего потенциометра записывает «нулевую» линию (рис. 3.5).
Хроматографическую колонку с исследуемым адсорбентом помещают в сосуд Дьюара с жидким азотом, продолжая пропускать через нее газовую смесь. При охлаждении колонки в результате адсорбции азота и изменения состава газовой смеси перо потенциометра начинает отклоняться от «нулевой» линии. Смещение пера потенциометра происходит до тех пор, пока сорбент полностью не будет насыщен азотом при данной концентрации азота в газовой смеси. Равновесие считается установленным, когда перо самописца снова отмечает «нулевую» линию. Затем сосуд с жидким азотом удаляют, и происходит полная десорбция азота с поверхности адсорбента при комнатной температуре. При этом самописец вычерчивает кривую десорбции (см. рис. 3.5). Операции адсорбции и десорбции при данном составе газовой смеси повторяют несколько раз (для оценки воспроизводимости результатов).
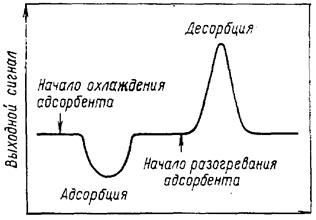
рисунок 3.5 – Хроматограмма адсорбции и десорбции азота.
Таблица 3.4 – Экспериментальные и расчетные данные по определению удельной поверхности адсорбента методом тепловой десорбции
| х, % (об.) | Р, Па | 
| A | 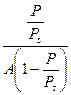
| A¥ |
Подобные операции проводят, меняя концентрацию азота в газовой смеси (через 5 %) в интервале 5 – 25 % (об.). Количество азота, адсорбированное при каждой исходной концентрации азота в газовой смеси, определяют по площади пика на кривой десорбции, используя калибровочный график.
Для получения калибровочного графика «объем азота (мл) – площадь пика» проводят хроматографический анализ модельных азотгелиевых смесей. Рассчитывают величину адсорбции А как отношение сорбированного азота к массе сорбента в колонке.
Парциальное давление азота р, при котором проводилась адсорбция, находят по формуле
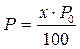 (3.32)
(3.32)
где х – содержание азота в газовой смеси, % (об.);
р0 – барометрическое давление.
Далее рассчитывают относительное давление азота (где ps – давление насыщенного пара азота). Строят изотерму адсорбции А – . Представляют изотерму в координатах линейной формы уравнения БЭТ:
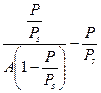
Отрезок, отсекаемый на оси ординат, соответствует величине 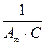 , а тангенс угла наклона этой прямой равен
, а тангенс угла наклона этой прямой равен 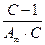 . Из этих данных рассчитывают значение А¥. Полученные результаты записывают в таблицу (см. табл. 3. 4).
. Из этих данных рассчитывают значение А¥. Полученные результаты записывают в таблицу (см. табл. 3. 4).
Найдя значение А¥, по уравнению (3.4) вычисляют удельную поверхность исследуемого адсорбента, приняв для азота s0 = 0,162 нм2.
Работа 21 «Хроматографическое разделение смеси ионов с помощью ионообменных смол»
Цель работы: определение полной обменной емкости катионита и константы ионного обмена; разделение смеси катионов на ионообменной хроматографической колонке.
Краткая теория.Ионный обмен представляет собой обратимое стехиометрическое замещение подвижного иона, связанного с ионогенной группой ионита, на другой одноименно заряженный ион, находящийся в растворе. Количественной характеристикой ионита является полная обменная емкость ПОЕ. Определение ПОЕ можно осуществить статическим или динамическим методом, основанном обычно на реакциях, протекающих в водных растворах:
RSO3–H+ + NaOH ® RSO3–Na+ + Н2О
RNH3+OH– +HCl ® RNH3+Cl– + Н2О
При статическом методе смолу, например катионит в Н+-форме, титруют раствором щелочи.
При динамическом методе ПОЕ определяется с помощью хромато-графических колонок. Через колонку, заполненную ионообменной смолой, пропускают раствор электролита и регистрируют зависимость концентрации поглощаемого иона в выходящем растворе (элюате) от объема прошедшего раствора (выходная кривая).
В работе ПОЕ сульфокатионита в Н+-форме определяют динамическим методом по количеству кислоты в элюате, образующейся в результате вытеснения из смолы ионов Н+ ионами Na+:
RSO3–H+ + NaCl ® RSO3–Na+ + Н+ + Cl–
ПОЕ рассчитывают по формуле
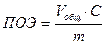 (3.33)
(3.33)
где Vобщ – суммарный объем раствора, содержащий вытесненную из смолы кислоту;
с – концентрация кислоты;
m – масса ионообменной смолы в колонке.
Константу ионного обмена можно определить из данных о равновесном распределении ионов в статических условиях (равновесное состояние при ионном обмене описывается законом действия масс), а также динамическим методом по скорости перемещения зоны вещества по слою смолы (элюентная хроматография).
Элюúрование– [< лат. eluere вымывать]– извлечение вещества вымыванием его подходящим растворителем – элюентом.
Если через колонку с катионитом, в верхней части которой находится сорбированный ион Мz+, пропускается раствор кислоты, то в смоле происходит многократный процесс обмена:
zRSO3–H+ + Мz+ Û (RSO3–)z Мz+ + zН+
В условии равновесия при распределении ионов между протекающим раствором и слоями ионита (равновесная хроматография) отношение между концентрациями иона Mz+ в смоле 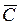 и растворе с (при малых значениях концентрации) равно
и растворе с (при малых значениях концентрации) равно
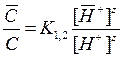 (3.34)
(3.34)
Скорость перемещения хроматографической зоны с постоянной концентрацией иона по высоте колонки равна
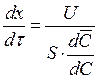 (3.35)
(3.35)
где U – объемная скорость пропускания раствора кислоты;
s – площадь сечения колонки.
В реальных условиях равновесие не успевает полностью установиться вследствие медленной диффузии ионов в зернах смолы, что приводит к размытию хроматографической зоны. Приведенное уравнение (3.35) хорошо описывает скорость движения зоны с максимально возможной концентрацией иона. Выражение (3.35) можно преобразовать следующим образом:
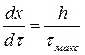 , Vмакс = U . tмакс,
, Vмакс = U . tмакс, 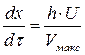 (3.36)
(3.36)
где Vмакс и tмакс – объем протекающего через колонку раствора и время, отвечающие максимуму выходной кривой;
h – длина колонки.
При малых концентрациях иона Мz+ и постоянной концентрации кислоты производную 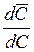 можно заменить на отношение конечных величин
можно заменить на отношение конечных величин 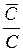 и считать концентрацию ионов водорода в смоле [Н+] равной ПОЕ. Тогда
и считать концентрацию ионов водорода в смоле [Н+] равной ПОЕ. Тогда
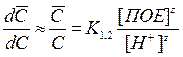 (3.37)
(3.37)
Из уравнений (3.35) – (3.37) следует:
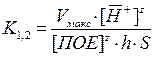 (3.38)
(3.38)
Для двухвалентного катиона М2+, например кобальта или меди, концентрационная константа равновесия

рассчитывается по уравнению (3.38) при z = 2. Значение Vмакс находят с учетом поправки на свободный объем колонки V0:
Vмакс = V*макс – V0 (3.39)
где V*макс – объем элюата, вышедшего из колонки от начала элюирования М2+ кислотой до появления максимальной концентрации М2+ в элюате (максимум на выходной кривой).
При хроматографическом разделении ионов широко используется различная их склонность к образованию комплексных соединений. Например, кобальт и медь могут быть разделены в колонке с сульфокатионитом при помощи десорбента – раствора цитрата калия.
Если пропустить через катионит в K+-форме раствор, содержащий небольшое количество разделяемых элементов в отсутствие комплексо-образователя, то ионы кобальта и меди поглощаются в верхнем слое смолы.
Разделить кобальт и медь динамическим методом, промывая колонку раствором, содержащим ионы K+, трудно, так как константы обмена ионов Со2+ и Сu2+ на ион K+ отличаются не намного.
Если через колонку пропускать раствор цитрата калия (десорбент), то при контакте его со смолой происходит частичная десорбция меди и кобальта вследствие комплексообразования, например:
(RSO3–)2 М2+ + 2K+ + А3– Û 2RSO3–K+ + [MA]–
где А3– – комплексообразующий ион лимонной кислоты.
С лимонной кислотой ионы кобальта и меди образуют несколько различных комплексных соединений [МА]–, [МА2]–4 и др., которые не адсорбируются на катионите.
Общая концентрация ионов меди или кобальта в растворе равна
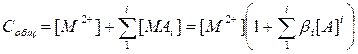 (3.40)
(3.40)
где bi – константы устойчивости комплексных ионов;
i – число комплексообразующих ионов в комплексном соединении.
Уравнение (3.35) для скорости перемещения вещества по колонке с учетом выражений (3.37) и (3.40) в присутствии комплексообразователя имеет вид
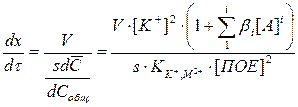 (3.41)
(3.41)
Из уравнений (3.40) и (3.41) следует, что в присутствии комплексообразователя уменьшается концентрация катионов, способных адсорбироваться на катионите, возрастает концентрация неадсорбирующихся комплексных соединений (при постоянном собщ) и в результате увеличивается скорость перемещения ионов данного вещества по колонке. При протекании вымывающего раствора через колонку происходит многократная десорбция и сорбция разделяемых ионов, причем катионы меди, образующие более устойчивые комплексные соединения, перемещаются вдоль слоя смолы с большей скоростью, чем ионы кобальта. В результате в колонке формируются различные по окраске зоны – для меди голубая, для кобальта оранжевая; первым из колонки выходит раствор, содержащий комплексные соединения меди, затем – кобальта (рис. 3.6).
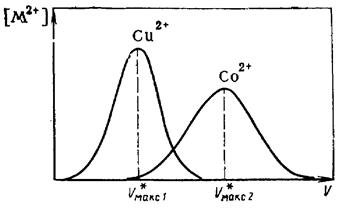
рисунок 3.6 – Выходные кривые хроматографического разделения ионов меди и кобальта.
Скорость перемещения вещества по колонке обратно пропорциональна Vмакс. Относительная скорость перемещения по колонке разделяемых ионов может быть рассчитана по уравнению
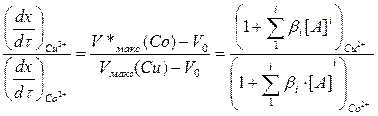 (3.42)
(3.42)
Поскольку константы ионного обмена K+ на Со2+ и Сu2+ приблизительно одинаковы ( 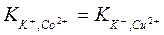 ), относительная скорость численно равна параметру, характеризующему относительную степень комплексообразования разделяемых ионов.
), относительная скорость численно равна параметру, характеризующему относительную степень комплексообразования разделяемых ионов.
Дата добавления: 2020-07-18; просмотров: 857;
Поиск по сайту
Узнать еще
- Cпецифика логопедической работы в остром периоде
- H. Разработка мер по повышению качества работы органа здравоохранения
- I. Определение условий выполнения рукописи.
- II раздел. Организация работы логопеда в группе для детей с ОНР
- II. Види виробничої документації та порядок її ведення
- II. Порядок разработки, утверждения, внесения изменений в Инструкцию по делопроизводству
- III. Порядок присвоения спортивных званий
- IV. Выполнение работы

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
