Стремление энтропии системы к максимальному значению (условие экстремума) указывает на приближение системы к равновесию.
Следовательно, в изолированной системе знак изменения энтропии является критерием направленности самопроизвольного процесса:
Если ΔS=0, то S=Smax; система находится в состоянии термодинамического равновесия (параметры состояния во времени не изменяются)
Если ΔS>0, тоS→Smax, (энтропия растет)- процесс самопроизвольно протекает в прямом направлении, т.е. термодинамически возможен.
Если ΔS<0, то S→Smin, (энтропия убывает)– самопроизвольно может развиваться только обратный процесс, прямой процесс т/д невозможен.
Только возрастанием энтропии можно объяснить самопроизвольное протекание в изолированных системах таких процессов, как передача теплоты от более нагретого тела к менее нагретому, или взаимная диффузия (смешение) двух или нескольких химически не взаимодействующих газов.
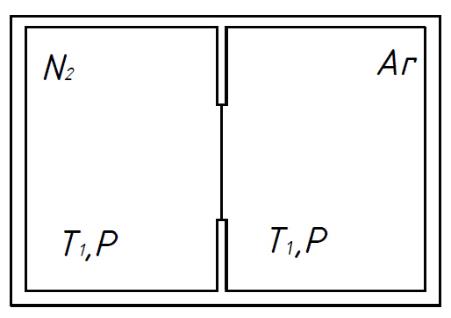
Объединим две различные изолированные системы в одну (состояние 1). Пусть р, Т, U, V = const, но нашелся «волшебник», который проник внутрь изолированной системы и разрушил перегородку.
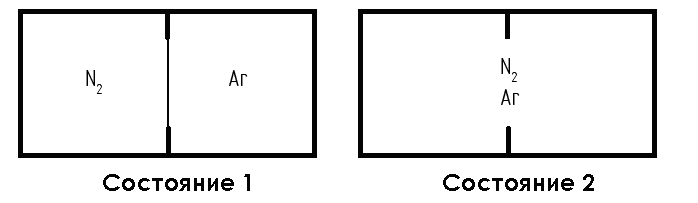 Схема смешения двух химически не взаимодействующих газов, процесс 1→2
Схема смешения двух химически не взаимодействующих газов, процесс 1→2
«Самопроизвольным, или спонтанным, является процесс, который совершается в системе без затраты работы извне и который уменьшает работоспособность системы после своего завершения»
Химическая термодинамика изучает системы, состоящие из большого числа частиц, неизбежно вступающие во взимодействие– макросистемы. В подобных системх различают так называемые макро- и микропараметры. Для каждой индивидуальной частицы характерно микросостояние, которое описывается микропараметрами (координаты в пространсте, векторы скорости, энергия). Совокупность микропараметров каждой частицы называется микросостоянием частицы.
К макрохарактеристикам относятся независимые переменные, свойственные системе в целом (давление (р), объем (V), температура (T), концентрации веществ (ci), которые определяются экспериментально или рассчитываются теоретически.
Для описания системы на таком уровне потребовалась новая характеристика – термодинамическая вероятность состояния частицы.
Термодинамическое вероятностное состояние – число микросостояний, посредством которых реализуется данное макросостояние системы. В отличие от математической вероятности, которая может принимать значения от нуля до единицы, термодинамическое вероятность представляет собой очень большую величину, дополнительно зависящую от числа частиц в системе: для системы из 10-ти частиц она имеет порядок 104. Она обозначается буквойWи изменяется в диапазоне [1; +∞). Связь энтропии с термодинамической вероятностью установил Л. Больцман, представив ее в виде уравнения, названного его именем:
S=k*lnW , (13)
где k= R / NА – константа Больцмана, равная 1,38*10-23Дж/К.
Всякая изолированная система самопроизвольно стремиться принять состояние, характеризующееся максимальной термодинамической вероятностью– наиболее общая формулировка второго начала термодинамики.
На основании уравнения Больцмана можно показать, что любой необратимый процесс, самопроизвольно протекающий в изолированной системе, характеризуется увеличением энтропии. Рассмотрим процесс 1→2, смешение двух химически не взаимодействующих газов в условиях, представленных на рисунке. В состоянии 1 термодинамическая вероятность системы равна W1. При удалении перегородки газы начнут самопроизвольно диффундировать друг в друга (смешиваться) до тех пор, пока молекулы каждого газа равномерно не распределятся по всему объему. В конечном состоянии 2 термодинамическая вероятность системы равна W2. Система самопроизвольно перешла из менее вероятного состояния в более вероятное (W2>W1) при отсутствии энергетического обмена с окружающей средой. Единственной причиной протекания этого процесса является изменение энтропии:
ΔS=S2-S1=k*lnW2- k*lnW1= k*ln(W2/W1);
Но W2>W1, W2/W1> 1, ln(W2/W1)>0, следовательно, ΔS>0;
На этой основе сформулирован второй закон термодинамики для изолированных систем:
В изолированных системах самопроизвольно могут совершаться только такие необратимые процессы, в которых энтропия возрастает, т.е. ΔS>0.
Значение энтропии является мерой неупорядоченности процесса, зависит от агрегатного состояния, T, P и сложности системы, природы вещества. При прочих равных условиях:
Sг>Sж>Sтв
Энтропия простых веществ зависит от их аллотропной формы:
S (Сграфит) >S(Салмаз) илиS (o2) <S (o3).
При повышении температуры энтропия возрастает:
если Т1>T2, тоS1>S2.
Энтропия системы при повышении давления уменьшается, так как снижается неупорядоченность движения частиц:
Если р1>р2 , тоS1<S2.
Энтропия системы при увеличении ее сложности повышается, т.к. вариантов расположения частиц становится больше.
Для энергетических характеристик вещества при стандартных условиях наряду со стандартной энтальпией Н0 используется стандартная энтропия S0вещества.
В отличие от стандартной энтальпии, стандартная энтропияпростых веществ не равна нулю. Энтропия всех веществ всегда больше нуля.
Все процессы, сопровождающиеся увеличением молекулярного беспорядка, в системе ведут к увеличению энтропии (ΔS>0):
- плавление и сублимация твердых веществ;
- испарение жидкостей;
-расширение газов;
- растворение кристаллов;
- химические реакции, сопровождающиеся увеличением количества вещества.
В процессах, связанных с затвердеванием жидкостей, кристаллизацией веществ из растворов, конденсацией паров и сжатием газов, степень молекулярного беспорядка уменьшается и энтропия системы убывает (∆S < 0).
Все перечисленные процессы мы обнаруживаем в разнообразных технологических цепочках, используемых при производстве композитов. Учет изменения энтропии системы в используемых процессах – необходимый элемент их научного обоснования.
Вычисление энтропии в различных процессах(обозначениеDпрS):
Пример 1. Фазовоепревращение чистого вещества.
При фазовых переходах первого рода энтропия и объем системы изменяются скачком, а температура остается постоянной (примеры: ). На практике эти переходы осуществляются обратимо в изобарно-изотермических условиях (p, T = const )по следующей схеме:
Фаза 1 ↔ Фаза 2; DН пр=
Изменение энтропии при фазовых переходах первого рода вычисляют, интегрируя уравнение (4) при p, T = const (Q = Qp):
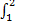 dS= 1 / T dQр
dS= 1 / T dQр
DпрS = S2 – S1 = Qp /T = DНпр /T, (14)
где DН пр, Дж/моль - это молярное изменение энтальпии (молярнаятеплота фазового перехода), Т – температура фазового перехода, К.
Пример 2. Изменение энтропии одного моля идеального газа в различных равновесных процессах:
Изменение энтропии одного моля идеального газа в различных равновесных процессах:
а) изохорный (V = const, dV = 0) нагрев от T1 до T2. Неравенство (9) для равновесного изохорного процесса превратится в уравнение
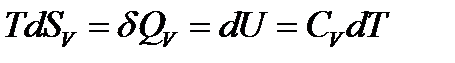
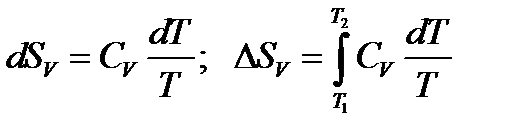
Если принять CV = const в интервале температур T1‑T2, то
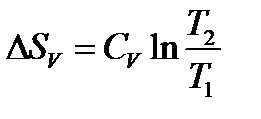 (15)
(15)
б) изобарный (p = const) нагрев от T1доT2:
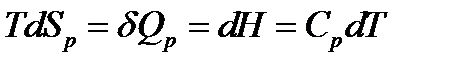
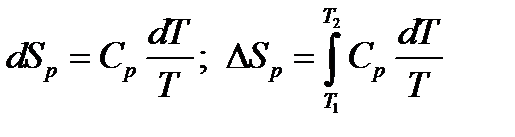
Принимая Cp = constв интервале температур T1‑T2, получаем
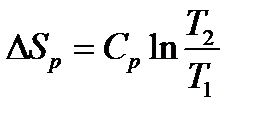 (16)
(16)
Поскольку зависимости внутренней энергии U = CVT (CV = f(T)) и энтальпии H = CpT (Cp = f(T)) от температуры справедливы для любого агрегатного состояния вещества, уравнения (15) и (16) можно использовать для расчета изменений энтропии в процессах изохорного и изобарного нагрева (и охлаждения) конденсированных веществ;
в) изотермическое (T = const) расширение объема от V1 до V2 (напомним, что для идеального газа при T = const, U = const, dU = 0):
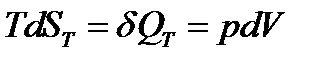
но для одного моля идеального газа 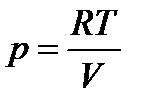 , тогда
, тогда
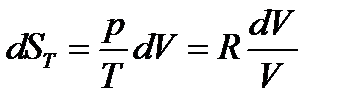
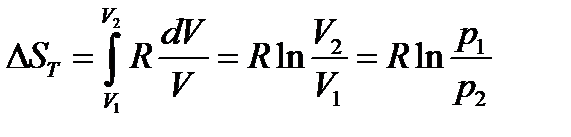 (17)
(17)
так как, согласно закону Бойля – Мариотта, (pV)T = const.
По формулам (14) – (17) вычисляются молярные изменения энтропий веществ в процессах (Дж/(моль·К)). Если количество вещества в системе равно n моль, то при умножении соответствующих молярных значений ΔS на n получают изменение энтропии системы в Дж/К.
Третий закон термодинамики
В отличие от абсолютных значений внутренней энергии (U) и энтальпии (Н), которые невозможно вычислить, абсолютные значения энтропии (S)поддаются определению как для сложных, так и для простых веществ из-за наличия у них начальной точки в шкале отсчета, установленной третьим законом термодинамики. Третье начало, называемое также постулатом Планка, звучит так: при температуре абсолютного нуля (Т=0К) энтропия идеальных кристаллов любого простого вещества или соединения равна нулю:limSТ → 0 = 0, или S0 = 0.Предполагается, что в чистом, не имеющем дефектов кристалле вещества существует абсолютный порядок в расположении частиц , приТ = 0 К возможно единственное состояние системы, при котором частицы«застывают» в узлах кристаллической решетки, термодинамическая вероятность равна минимальному значению (W=1), поэтому для одного моля вещества S0 = RlnW = Rln1 = 0.
Для неидеальных кристаллов, смесей, твердых растворов, стекол всегда S0 >0, а W>1, т.е. у таких веществ существует так называемая нулевая энтропия, связанная с дефектами кристаллической решетки и др. причинами.
Стандартное значение энтропии обозначается символом S2980, Дж/(моль· К). Стандартные значения энтропий S2980различных веществ приводятся в таблицах термодинамических величин; это всегда положительная величина. Изменения энтропии в процессах (DS) могут быть положительными, отрицательными или равными нулю.
Схема расчета стандартных энтропий S2980
Вблизи абсолютного нуля все вещества находятся в твердом состоянии. Если при стандартных термодинамических условиях вещество находится в кристаллическом состоянии, то в интервале температур 0 – 298 К энтропия изменяется только за счет повышения температуры, следовательно в соответствии с (16) при постоянном давлении
dS = dQр /T= dН / T = CрdT /T,
или 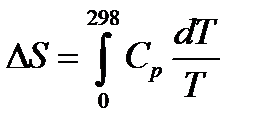 ,значит
,значит 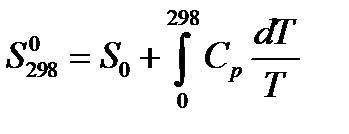 , ноS0 = 0в соответствии с третьим началом, таким образом,
, ноS0 = 0в соответствии с третьим началом, таким образом,
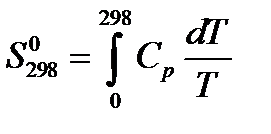
Если при Т =298 К вещество является газом, надо учесть изменения энтропии, связанные с фазовыми переходами (плавление, испарение):
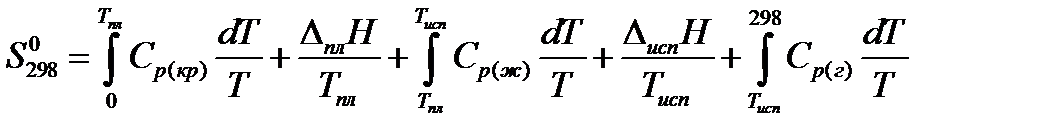 (21)
(21)
Если при T = 298К вещество находится в жидком состоянии, при расчете стандартного значения энтропии в формуле (21) ограничиваются тремя первыми слагаемыми. Уравнение (21) является общим математическим выражением для расчета стандартных значений энтропий.
Так как энтропия является функцией состояния системы, то стандартное изменение энтропии в результате осуществления химической реакции (стандартная энтропия реакции) вычисляется как разность между суммой стандартных энтропий продуктов реакции и суммой стандартных энтропий исходных веществ с учетом стехиометрии процесса:
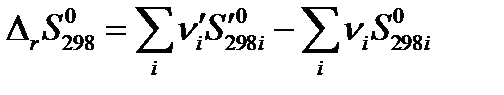 (22)
(22)
т.е.
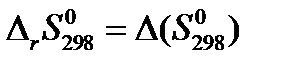 (23)
(23)
где Δ в правой части уравнения (23) – знак оператора.
Лекция. Изменение энергии Гиббса и Гельмгольца – критерии направленности самопроизвольного процесса в закрытой системе
Реальные процессы проводятся, как правило, в закрытых системах в изобарно-изотермических (р, Т = соnst) или изохорно-изотермических (V, Т = соnst) условиях.
Для неизолированных систем нужно учитывать не только изменение энтропии, но и изменение энергии. Поэтому необходимо рассматривать две тенденции, определяющие направление самопроизвольно протекающих процессов:
1) стремление системы к достижению минимума энергии;
2) стремление системы к максимуму энтропии, т.е. к неупорядоченности.
Все процессы, при которых энергия в системе уменьшается, а энтропия возрастает, протекают самопроизвольно. Самопроизвольность других процессов зависит от того, какая из этих двух тенденций – энергетическая или энтропийная – окажется более эффективной, какая из этих противоборствующих тенденций получит перевес над другой. В этом проявляется противоречивость материального мира.
Для однозначной формулировки условия протекания самопроизвольных процессов в любых системах оказалось необходимым ввести еще две термодинамические функции, которые характеризовали бы одновременно и энергетику, и неупорядоченность данных систем. Впервые такую термодинамическую функцию ввел Д.У. Гиббс, и в память об этом выдающемся американском ученом ее назвали энергией Гиббса.
Критерием направленности самопроизвольного процесса в этих случаях является не знак изменения энтропии (ΔS), а знак изменения энергии Гиббса (ΔG) или энергии Гельмгольца (ΔF) в системе.
За энергию Гиббса (изобарно-изотермический потенциал) и энергию Гельмгольца (изохорно-изотермический потенциал) по рекомендации Международного союза теоретической и прикладной химии (IUPAC) принимаются следующие выражения:
Дата добавления: 2021-02-19; просмотров: 576;
Поиск по сайту
Узнать еще
- F45.38 другие органы или системы
- I. История возникновения и развития классно-урочной системы.
- I. Развитие Донбасса в условиях кризиса феодально-крепостнической системы
- I. Создание системы управленческого учета.
- III. Филогенез эндокринной системы.
- IV. Движение поездов при неисправности электрожезловой системы и порядок регулировки количества жезлов в жезловых аппаратах
- L.3. Состояние российской системы образования и необходимость ее
- s, p - ПРИБЛИЖЕНИЕ, p-ЭЛЕКТРОННЫЕ МЕТОДЫ

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
