Синдром Прадера-Вилли
Впервые синдром Прадера-Вилли был описан в 1956 году. Причиной возникновения этого синдрома является потеря функции хромосомных участков, расположенных в проксимальной части длинного плеча хромосомы 15 (15q11-13). Делеция имеет отцовское происхождение и наблюдается у 70% больных, у 5% заболевание связано с перестройкой хромосомы 15. В большинство случаев заболевание возникает de novo, в 25% случаев синдром возникает в результате однородительской дисомии. У некоторых больных хромосомную аномалию не удается идентифицировать, но у них наблюдается характерная клиническая картина синдрома Прадера-Вилли.
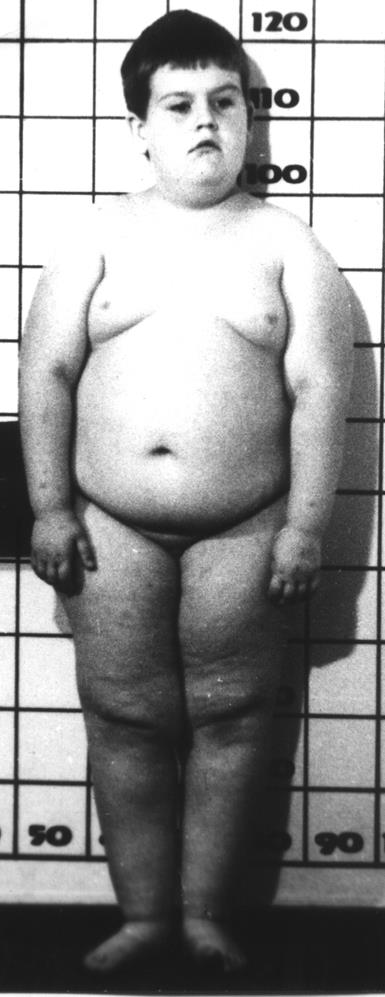
4.5.9
Основными клиническими признаками являются отставание умственного развития, неадекватное поведение, задержка физического развития, низкорослость, гипотония. Одни клинические признаки при этом заболевании можно наблюдать до 3 летнего возраста (мышечная гипотония, малый вес и трудности вскармливания), другие начинают преобладать после 6 месячного возраста (ожирение, усиление аппетита, нарастание умственной отсталости, отставание в росте). Наряду с диспластическими признаками (опущенные углы рта, высокое небо, гипертелоризм, эпикант, маленькие стопы и кисти, миндалевидный разрез глаз, аномалии дерматоглифика) выявляется гипогонадизм, обусловленный низким уровнем половых гормонов, гипопигментация (у75% больных). Следует отметить, что синдром Прадера-Вилли характеризуется широким клиническим полиморфизмом, поэтому необходимо проводить дифференциальную диагностику с синдромами Коэна, Опица-Фриаса, Барде-Бидля.
Продолжительность жизни составляет 25 – 30 лет.
Диагностика заболевания осуществляется с помощью ДНК-анализа или методом FISH. Риск для сибсов пробанда – около 1%.
Синдром Ангельмана
Если для возникновения синдрома Прадера-Вилли основной причиной являлась делеция проксимальной части длинного плеча хромосомы 15 отцовского происхождения, то аналогичная потеря той же части длинного плеча хромосомы 15, но только материнского происхождения обусловливает развитие другой патологии – синдрома Ангельмана. При этом заболевании развивается совсем другая клиническая картина. Для синдрома Ангельмана характерно: выраженная олигофрения, задержка речи, гиперактивное поведение, судороги, большая нижняя челюсть, макростомия, гипопигментация (у 40% больных). Они поздно начинают ходить, для них характерна походка с широко расставленными ногами, локтевые суставы согнуты; отмечается насильственный немотивированный смех, имеются выраженные расстройства координации движений. 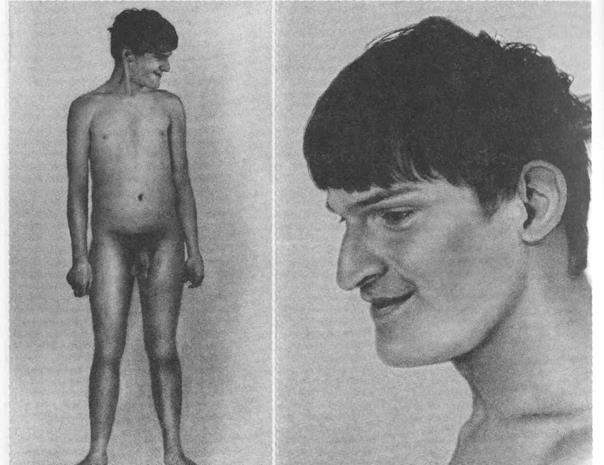
4.5.10
Дифференциальную диагностику следует проводить с синдромами Петерса-Пласа, Ретта и с тригоноцефалией Опица.
Частота синдрома в популяции составляет 1:20 000.
Примерно 20 – 30% больных не имеют делеции проксимальной части длинного плеча хромосомы 15; у незначительного числа больных причиной является однородительская дисомия. Диагностика синдрома осуществляется теми же методами, что и при синдроме Прадера-Вилли, т.е. проводится ДНК-анализ и метод FISH. С помощью этих методов можно установить этиологию около 90% случаев заболевания. Риск для сибсов пробанда не известен.
Дата добавления: 2016-10-26; просмотров: 2654;
Поиск по сайту
Узнать еще
- I. РАЗДЕЛ ПО ПРОБЛЕМЕ НЕДОСТАТОЧНОСТИ МИТРАЛЬНОГО КЛАПАНА (СИНДРОМ МИТРАЛЬНОЙ РЕГУРГИТАЦИИ)
- II. РАЗДЕЛ ПО ПРОБЛЕМЕ СТЕНОЗА МИТРАЛЬНОГО ОТВЕРСТИЯ ( СИНДРОМ МИТРАЛЬНОЙ ОБСТРУКЦИИ ).
- А Нейропсихологические синдромы поражения задних отделов коры больших полушарий головного мозга.
- Абеталипопротеинемия (синдром Бассена-Корнцвейга)
- Абстинентный синдром
- Альтернирующие синдромы при поражении ствола головного мозга
- АНТИФОСФОЛИПИДНЫЙ СИНДРОМ
- Антифосфолипидный синдром и волчаночный антикоагулянт

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
