Степени окисления: 6, 5, 4, 3, 2
Главным природным соединением молибдена является молибденит, или молибденовый блеск: MоS2 — минерал, очень похожий по внешнему виду на графит и долгое время считавшийся таковым.
При обработке молибденового блеска азотной кислотой получается белый остаток, обладающий свойствами кислоты. Назвали его молибденовой кислотой и сделал заключение, что сам минерал представляет собой сульфид нового элемента. Пять лет спустя этот элемент был получен в свободном состоянии путем прокаливания молибденовой кислоты с древесным углем.
Для получения металлического молибдена из молибденового блеска последний переводят обжигом в МоО3, из которого металл восстанавливают водородом. При этом молибден получается в виде порошка.
Молибден — серебристо-белый металл плотностью 10,2 г/см3, плавящийся при 2620 °С. При комнатной температуре он не изменяется на воздухе, но при накаливании окисляется в белый триоксид МоО3.
С водородом Молибден химически не реагирует вплоть до плавления. Фтор действует на Молибден при обычной температуре, хлор при 250 °С, образуя MoF6 и МоСl6. При действии паров серы и сероводорода соответственно выше 440 и 800 °С образуется дисульфид MoS2. С азотом Молибден выше 1500 °С образует нитрид (вероятно, Mo2N). Твердый углерод и углеводороды, а также оксид углерода (II) при 1100-1200 °С взаимодействуют с металлом с образованием карбида Мо2С (плавится с разложением при 2400 °С). Выше 1200 °С Молибден реагирует с кремнием, образуя силицид MoSi2, обладающий высокой устойчивостью на воздухе вплоть до 1500-1600 °С
Достоверно установлено существование трех сульфидов Молибдена - МоS3, MoS2 и Mo2S3.
Кроме того, известны промежуточные оксиды, соответствующие по составу гомологическому ряду МоnO3n-1 (Мо9О26, Мо8О23, Мо4О11); все они термически неустойчивы и выше 700 °С разлагаются с образованием МоО3 и МоО2.
Оксид МоО3 образует простые (или нормальные) кислоты Молибдена - моногидрат Н2МоО4, дигидрат Н2МоО4·Н2О и изополикислоты - H6Mo7O24, HМo6O24, H4Мo8O26 и другие. Соли нормальной кислоты называется нормальными молибдатами, а поликислот - полимолибдатами.
Одна из распространенных солей гетерополикислот - фосфоромолибдат аммония (NH4)3[Р(Мо3О10)4]·6Н2О
Соляная и разбавленная серная кислоты при комнатной температуре не действуют на молибден; он растворяется в азотной кислоте или горячей концентрированной серной кислоте. Эти кислоты растворяют защитную оксидную пленку на поверхности металла. В результате молибден окисляется до молибденовой кислоты Н3Мо04.
Около 80% всего добываемого молибдена расходуется на производство специальных сортов стали. Он входит в состав многих нержавеющих сталей; кроме того, его введение способствует увеличению их жаропрочности.
В своих соединениях молибден проявляет положительные степени окисления: шесть, пять, четыре, три и два. Наиболее стойкими являются соединения молибдена (VI). Важнейшие из них — соли молибденовой кислоты Н2МоО4 (молибдаты), часто имеющие сложный состав.
Молибдат аммония (NH4)6Mo24*4H2O применяется в анализе для открытия и количественного определения фосфорной кислоты, с которой он образует характерный желтый осадок состава (NH4)3PO4*12Mo3*6H2O. Последняя пред-ставляет собой аммонийную соль комплексной фосфорномолибденовой кислоты, относящейся к классу гетерополикислот.
Вольфрам
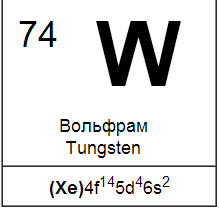
По распространенности в земной коре вольфрам уступает хрому, но превосходит молибден. Природные соединения вольфрама в большинстве случаев представляют собой вольфраматы - соли вольфрамовой кислоты H2WO4. Так, важнейшая вольфрамовая руда — вольфрамит — состоит из вольфраматов железа и марганца. Часто встречается также минерал шеелит CaWO4.
Для выделения вольфрама из вольфрамита последний сплавляют в присутствии воздуха с содой. Вольфрам переходит в вольфрамат натрия Na2W04, который извлекают из полученного сплава водой, а железо и марганец превращаются в нерастворимые соединения Fе2О3 и Мn304.
Из водного раствора действием соляной кислоты выделяют свободную вольфрамовую кислоту в виде аморфного желтого осадка:
Na2W04 + 2НС1 = H2W04 +2NaCl.
При прокаливании вольфрамовая кислота переходит в триоксид вольфрама WO3. Восстанавливая его водородом или углеродом (для чего используют чистые сорта сажи), получают порошок металлического вольфрама, подвергаемый в дальнейшем для получения компактного металла такой же обработке, как и порошок молибдена
Вольфрамовому ангидриду соответствует вольфрамовая кислота H2WO4 - желтый порошок, практически не растворимый в воде и в кислотах. При ее взаимодействии с растворами щелочей и аммиака образуются растворы вольфраматов. При 188°С Н2WО4 отщепляет воду с образованием WO3.
Галогены, сера, углерод, кремний, бор взаимодействуют с Вольфрамом при высоких температурах (фтор с порошкообразным Вольфрамом - при комнатной). С водородом Вольфрам не реагирует вплоть до температуры плавления; с азотом выше 1500°С образует нитрид.
В соединениях Вольфрам проявляет валентность от 2 до 6, наиболее устойчивы соединения высшей валентности.
Вольфрам образует четыре оксида: высший - WO3 (вольфрамовый ангидрид), низший - WO2 и два промежуточных W10О29 и W4O11
Наиболее важные из них: WCl6 (tпл 275°С, tкип 348°C) и WO2Cl2 (tпл 266°С, выше 300°С сублимирует), получаются при действии хлора на вольфрамовый ангидрид в присутствии угля.
С серой Вольфрам образует два сульфида WS2 и WS3. Карбиды вольфрама WC (tпл2900°C) и W2C (tпл 2750°С) - твердые тугоплавкие соединения; получаются при взаимодействии Вольфрама с углеродом при 1000-1500°С.
Вольфрам — тяжелый белый металл плотностью 19,3 г/см3. Его температура плавления (около 3400 °С) выше, чем температура плавления всех других металлов. Вольфрам можно сваривать и вытягивать в тонкие нити.
На воздухе вольфрам окисляется только при температуре красного каления. Он очень стоек по отношению к кислотам, даже к царской водке, но растворяется в смеси азотной кислоты и фтороводорода:
W + 2HN03 + 8HF = H2[WF8] +2NO + 4Н20.
октафторовольфрамовая (VI) кислота
Большая часть добываемого вольфрама расходуется в металлургии для приготовления специальных сталей и сплавов. Быстрорежущая инструментальная сталь содержит до 20% вольфрама и обладает способностью самозакаливаться. Такая сталь не теряет своей твердости даже при нагревании докрасна. Поэтому применение резцов, сделанных из вольфрамовой стали, позволяет значительно увеличить скорость резания металлов.
Карбид вольфрама WC обладает очень высокой твердостью (близкой к твердости алмаза), износоустойчивостью и тугоплавкостью. На основе этого вещества созданы самые производительные инструментальные твердые сплавы. В их состав входит 85—95% WC и 5—15% кобальта, придающего сплаву необходимую прочность. Некоторые сорта таких сплавов содержат кроме карбида вольфрама карбиды титана, тантала и ниобия. Все эти сплавы получают методами порошковой металлургии и применяют главным образом для изготовления рабочих частей режущих и буровых инструментов: насадки резцов, сверл, фрез для обработки высокоуглеродистых и нержавеющих сталей. Однако при высоких температурах карбид состава WC разлагается с образованием другого, но менее твердого карбида вольфрама:
WC —> W2C + C.
При этом режущий инструмент разрушается. Поэтому для сохранности режущего инструмента важно его своевременное и достаточное охлаждение.
Соединения вольфрама очень сходны с соединениями молибдена. Из них наибольшее значение имеют вольфрамовая кислота H2WO4 и ее соли.
Билет 9.
Химия железа
Нахождение в природе. Встречается железо в виде различных соединений: оксидов, сульфидов, силикатов. В свободном состоянии железо находят только в метеоритах.
К важнейшим рудам железа относятся магнитный железняк Fe3O4, красный железняк Fe2О3, бурый железняк 2Fе203 • ЗН2О и шпатовый железняк FeCO3, железный колчедан, FeS2 (служит исходным сырьем для получения серной кислоты).
Физические и химические свойства железа. Соединения железа.
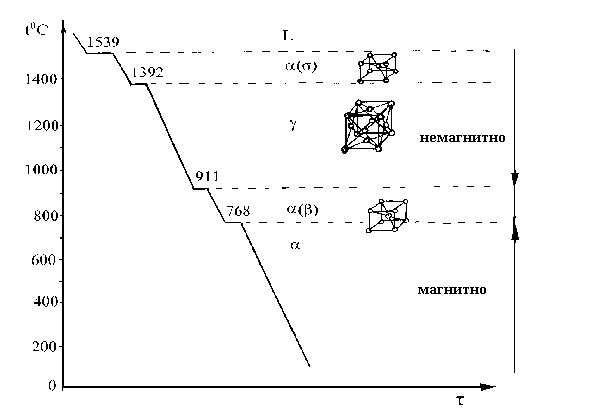
Температура плавления железа равна 1539±5 °С. Железо образует четыре кристаллические модификации: 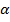 -,
-, 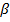 -,
-, 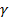 - и
- и 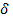 - железо. : -, -, и
- железо. : -, -, и 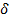 -железо имеют кубическую объемноцентрированную решетку с увеличивающимся расстоянием между ближайшими атомами железа ребра куба — элементарной ячейки от 286 пм через 290 пм до 293 пм, соответственно. Кубическую гранецентрированную решетку имеет 7-железо. Параметр кристаллической решетки 7-железа больше, чем параметры остальных модификаций, — 356 пм.
-железо имеют кубическую объемноцентрированную решетку с увеличивающимся расстоянием между ближайшими атомами железа ребра куба — элементарной ячейки от 286 пм через 290 пм до 293 пм, соответственно. Кубическую гранецентрированную решетку имеет 7-железо. Параметр кристаллической решетки 7-железа больше, чем параметры остальных модификаций, — 356 пм.
Температуры фазовых превращений железа хорошо видны на кривой охлаждения в виде остановок — горизонтальных площадок. Как видно, имеются площадки, отвечающие точкам перехода модификаций α-Fe 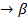 -Fe (768 °С), β -Fe —> β -Fe (912 °С), γ -Fe
-Fe (768 °С), β -Fe —> β -Fe (912 °С), γ -Fe 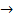 δ -Fe (1394°C). Температура 768 °C связана не только с изменением параметра объемноцентрированной кубической решетки, но и с изменением магнитных свойств железа. При температурах выше 768 °С железо парамагнитно, а ниже 768 °С — ферромагнитно.
δ -Fe (1394°C). Температура 768 °C связана не только с изменением параметра объемноцентрированной кубической решетки, но и с изменением магнитных свойств железа. При температурах выше 768 °С железо парамагнитно, а ниже 768 °С — ферромагнитно.
Чистое железо получают различными методами. Наибольшее значение имеют метод термического разложения пентакарбонила железа и электролиз водных растворов его солей.
Во влажном воздухе железо быстро ржавеет, т. е. покрывается бурым налетом гидратированного оксида железа, который вследствие своей рыхлости не защищает железо от дальнейшего окисления. В воде железо интенсивно корродирует; при обильном доступе кислорода образуются гидратные формы оксида железа (III):
2Fe + 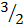 02 + nН20 = Fe203 * nН20.
02 + nН20 = Fe203 * nН20.
При недостатке кислорода или при его затрудненном доступе образуется Fe304:
3Fe + 202 + nН20 = Fe304 * nН20.
Железо растворяется в соляной кислоте любой концентрации:
Fe + 2НС1 = FeCl2 + Н2
Аналогично происходит растворение в разбавленной серной кислоте:
Fe + H2S04 = FeS04 + Н2
В концентрированных растворах серной кислоты железо окисляется до железа (III):
2Fe + 6H2S04 = Fe2(S04)3 + 3S02 + 6H20.
Однако в серной кислоте, концентрация которой близка к 100%, железо становится пассивным и взаимодействия практически не происходит.
В разбавленных и умеренно концентрированных растворах азотной кислоты железо растворяется:
Fe + 4HN03 = Fe(N03)3 + NO + 2H20 .
При высоких концентрациях HN03 растворение замедляется и железо становится пассивным.
Для железа характерны два ряда соединений: соединения железа (II) и соединения железа (III). Первые отвечают оксиду железа (II), FeO, вторые — оксиду железа (III), Fe203. Кроме того, известны соли железной кислоты H2Fe04, в которой степень окисления железа равна +6.
Соединения железа (II). Соли железа (II) образуются при растворении железа в разбавленных кислотах, в том числе очень разбавленной азотной кислоте. Важнейшая из них — сульфат железа (II), или железный купорос, FeS04 * 7Н20, образующий светло-зеленые кристаллы, хорошо растворимые в воде. На воздухе железный купорос постепенно выветривается и одновременно окисляется с поверхности, переходя в желто-бурую основную соль железа (III).
Сульфат железа (II) получают путем растворения обрезков стали в 20— 30%-ной серной кислоте:
Fe + H2S04 = FeS04 + Н2
При нагревании железного купороса выделяется вода и получается белая масса безводной соли FeS04. При температурах выше 480° С безводная соль разлагается с выделением диоксида и триоксида серы; последний во влажном воздухе образует тяжелые белые пары серной кислоты:
2FeS04 = Fe203 + S02 + S03
При взаимодействии раствора соли железа (II) со щелочью выпадает белый осадок гидроксида железа (II) Fe(OH)2, который на воздухе вследствие окисления быстро принимает зеленоватую, а затем бурую окраску, переходя в гидроксид железа (III) Fe(OH)3:
4Fe(OH)2 + 02 + 2Н20 = 4Fe(OH)3 .
Безводный оксид железа (II) FeO можно получить в виде черного легко окисляющегося порошка восстановлением оксида железа (III) оксидом углерода (II) при 500 °С:
Fe203 + СО = 2FeO + С02 .
Карбонаты щелочных металлов осаждают из растворов солей железа (II) белый карбонат железа(II) FeC03. При действии воды, содержащей С02, карбонат железа, подобно карбонату кальция, частично переходит в более растворимую кислую соль Fe(HC03)2. В виде этой соли железо содержится в природных железистых водах.
Соли железа (II) легко могут быть переведены в соли железа (III) действием различных окислителей азотной кислоты, перманганата калия, хлора, напри-мер:
6FeS04 + 2HN03 + 3H2S04 = 3Fe2(S04)3 + 2N0 + 4H20 ,
10FeS04 + 2KMn04 + 8H2S04 = 5Fe2(S04)3 + K2S04 + 2MnS04 + 8H20 .
Ввиду способности легко окисляться, соли железа (II) часто применяются как восстанови тели.
Соединения железа (III). Хлорид железа (III) FeCl3 представляет собой темно-коричневые с зеленоватым отливом кристаллы. Это вещество сильно гигроскопично; поглощая влагу из воздуха, оно превращается в кристаллогидраты, содержащие различное количество воды и расплывающиеся на воздухе. В таком состоянии хлорид железа (III) имеет буро-оранжевую окраску. В разбавленном растворе FeCl3 гидролизуется до основных солей. В парах хлорид железа (III) имеет структуру, аналогичную структуре хлорида алюминия и отвечающую формуле Fe2Cl6; заметная диссоциация Fe2Cl6 на молекулы FeCl3 начинается при температурах около 500 °С.
Сульфат железа (III) Fe2(S04)3 — очень гигроскопичные, расплывающиеся на воздухе белые кристаллы. Образует кристаллогидрат Fe2(S04)3 • 9Н20 (желтые кристаллы). В водных растворах сульфат железа (III) сильно гидролизован. С сульфатами щелочных металлов и аммония он образует двойные соли — квасцы, например железоаммонийные квасцы (NH4)Fe(S04)2*12H20 — хорошо растворимые в воде светло-фиолетовые кристаллы. При прокаливании выше 500° С сульфат железа (III) разлагается в соответствии с уравнением:
Fe2(S04)3 = Fe203 + 3S03
При действии щелочей на растворы солей железа (III) выпадает красно-бурый гидроксид железа (III) Fe(OH)3, нерастворимый в избытке щелочи.
Гидроксид железа (III) — более слабое основание, чем гидроксид железа (II); это выражается в том, что соли железа (III) сильно гидролизуются, а со слабыми кислотами (например, с угольной, сероводородной) Fe(OH)3 солей не образует. Гидролизом объясняется и цвет растворов солей железа (III): несмотря на то, что ион Fe3+ почти бесцветен, содержащие его растворы окрашены в желтобурый цвет, что объясняется присутствием гидроксо-ионов железа или молекул Fe(OH)3, которые образуются благодаря гидролизу:
Fe3+ + Н20 i  FeOH2+ + Н+;
FeOH2+ + Н+;
FeOH2+ + Н20 <=> Fe(OH)2 + +Н+;
Fe(OH)2 + + Н20 ó Fe(OH)3 + Н+.
При нагревании окраска темнеет, а при прибавлении кислот становится более светлой вследствие подавления гидролиза. При прокаливании гидроксид железа (III), теряя воду, переходит в оксид железа (III) Fe203. Оксид железа (III) встречается в природе в виде красного железняка и применяется как коричневая краска — железный сурик, или мумия
Характерной реакцией, отличающей соли железа (III) от солей железа (II), служит действие роданида каkbя KSCN или роданида аммония NH4SCN на соли железа. Раствор роданида калия содержит бесцветные ионы SCN-, которые соединяются с ионами Fe(III), образуя кроваво-красный, слабо диссоциирован-ный роданид железа (III) Fe(SCN)3. При взаимодействии же с роданидами ионов железа (II) раствор остается бесцветным.
Ферриты. При сплавлении оксида железа (III) с карбонатами натрия или калия образуются ферриты — соли не полученной в свободном состоянии железистой кислоты НFеО2, например феррит натрия NaFe02:
Fe203 + Na2CO3 = 2NaFe02 + C02
Образование ферритов указывает на амфотертность гидроксида железа (III). Ферриты так же получают сплавлением Fe(ОН)3 с щелочами или основными оксидами.
В технике ферритами или ферритными материалами называют продукты спекания порошков оксида железа (III) и оксидов некоторых двухвалентных металлов, например никеля, цинка, марганца. Ферриты обладают ценными магнитными свойствами и высоким электрическим сопротивлением, что обусловливает небольшую величину электрических потерь в них.
Ферромагнитный черный минерал магнетит Fe304, который иногда называют смешанным оксидом Fe0*Fe203, следует рассматривать как производное железистой кислоты и гидроксида железа (II). Это — соль Fe+2(Fe+302)2 — феррат (III) железа (II) или феррит железа (II).
Цианистые соединения железа. При действии на растворы солей железа (II) растворимых цианидов, например цианида калия, получается белый осадок цианида железа (II).
Fe2+ + 2CN- = Fe(CN)2
В избытке цианида калия осадок растворяется вследствие образования комплексной соли K4[Fe(CN)6] — гексацианоферрата (II) калия
Fe(CN)2 + 4KCN = K4[Fe(CN)6] или Fe(CN)2 + 4CN- = [Fe(CN)6]4-
С солями железа (III) K3[Fe(CN)6] образует зеленовато-бурый раствор.
В большинстве других комплексных соединений, как и в рассмотренных цианоферратах, координационное число железа (II) и железа (III) равно шести.
Соединения железа (VI). Если нагревать стальные опилки или оксид железа (III) с нитратом и гидроксидом калия, то образуется сплав, содержащий феррат калия K2Fe04 — соль железной кислоты H2Fe04:
Fe203 + 4К0Н + 3KN03 = 2K2Fe04 + 3KN02 + 2Н20 .
При растворении сплава в воде получается красно-фиолетовый раствор, из которого действием хлорида бария можно осадить нерастворимый в воде феррат бария BaFe04. Все ферраты — очень сильные окислители (более сильные, чем перманганаты). Соответствующая ферратам железная кислота H2Fe04 и ее ангидрид FeО3 в свободном состоянии не получены.
Карбонилы железа. Железо образует летучие соединения с оксидом углерода, называемые карбонилами железа. Пентакарбонил железа Fe(CO)5, представляет собой бледно-желтую жидкость, кипящую при 105 °С, нерастворимую в воде, но растворимую во многих органических растворителях. Fe(CO)5 получают пропусканием СО над порошком железа при 150—200 °С и давлении 10 МПа. Примеси, содержащиеся в железе, не вступают в реакции с СО, вследствие чего получается весьма чистый продукт. При нагревании в вакууме пентакарбонил железа разлагается на железо и СО.
Билет 10.
Химия марганца.
Из соединений, содержащих марганец, наиболее часто встречается минерал пиролюзит, представляющий собой диоксид марганца Мn02. Большое значение имеют также минералы гаусманит, Мn3О4 и браунит Мn2О3.
Марганец получают либо электролизом раствора MnS04, либо восстановлением из его оксидов кремнием в электрических печах. При электролитическом методе руду восстанавливают до соединений марганца со степенью окисления +2, а затем растворяют в смеси серной кислоты с сульфатом аммония. Получающийся раствор подвергают электролизу. Снятые с катодов осадки металла переплавляют в слитки.
Марганец — серебристо-белый твердый хрупкий металл. Известны четыре кристаллические модификации марганца, каждая из которых термодинамически устойчива в определенном интервале температур. Ниже 707 °С устойчив -марганец, имеющий сложную структуру —- в его элементарную ячейку входят 58 атомов. Сложность структуры марганца при температурах ниже 707 °С обусловливает его хрупкость. Хрупкость — следствие того, что в структуре марганца при понижении температуры все в большей мере проявляется ковалентная составляющая химической связи с участием его d-электронов.
В ряду напряжений марганец находится между алюминием и цинком.
На воздухе марганец покрывается тонкой оксидной пленкой, предохраняющей его от дальнейшего окисления даже при нагревании. Но в мелкораздробленном состоянии марганец окисляется довольно легко.
Вода при комнатной температуре действует на марганец очень медленно, при нагревании — быстрее. Он растворяется в разбавленных соляной и азотной кислотах, а также в горячей концентрированной серной кислоте (в холодной H2SO4 он практически нерастворим); при этом образуются катионы Мn2+.
Марганец образует четыре простых оксида (МnО, Мn203, МnО2 и Мn2О7) и смешанный оксид Мn3О4 (или МnО*Мn2О3). Первые два оксида обладают основными свойствами, диоксид марганца Мn02 амфотерен, а высший оксид Мn2О7 является ангидридом марганцовой кислоты НМnО4. Известны также производные марганца (VI), но соответствующий оксид МnО3 не получен.
В практическом отношении наиболее важны соединения марганца (II), диоксид марганца и соли марганцовой кислоты перманганаты, в которых марганец находится в степени окисления +7.
Соединения марганца (II). Соли марганца (II) получаются при растворении марганца в разбавленных кислотах или при действии кислот на различные природные соединения марганца. Так, из раствора, остающегося после получения хлора действием соляной кислоты на диоксид марганца, выкристаллизовывается хлорид марганца (II) МnС12 в виде бледно-розовых кристаллов. В твердом виде соли марганца (II) обычно розового цвета, растворы же их почти бесцветны.
При действии щелочей на растворы солей марганца (II) выпадает белый осадок - гидроксид марганца (II) Мn(ОН)2. Осадок легко растворяется в кислотах. На воздухе он быстро темнеет, окисляясь в бурый гидроксид марганца (IV) Мn(ОН)4
Оксид марганца (II) МnО получается в виде зеленого порошка при восстан-влении других оксидов марганца водородом.
Соединения марганца (IV). Наиболее стойким соединением марганца является темно-бурый диоксид марганца Мn02; он легко образуется как при окислении низших, так и при восстановлении высших соединений марганца. Как уже указывалось, Мn02 — амфотерный оксид; однако и кислотные, и основные свойства выражены у него очень слабо. Диоксид марганца широко применяется в качестве окислителя в химических источниках тока.
В кислой среде диоксид марганца — довольно энергичный окислитель. В качестве окислителя его применяют при получении хлора из соляной кислоты и в сухих гальванических элементах. Соли марганца (IV), например МnС14 и Mn(SO4)2, весьма нестойки.
Соединения марганца (VI) и марганца (VII). При сплавлении диоксида марганца с карбонатом и нитратом калия получается зеленый сплав, растворяющийся в воде с образованием красивого зеленого раствора. Из этого раствора можно выделить темно-зеленые кристаллы манганата калия К2Мn04 — соли марганцовистой кислоты Н2Мn04, очень нестойкой даже в растворе.
Реакцию образования манганата калия можно выразить уравнением:
Мn02 + К2С03 + KN03 = К2Мn04 + KN02 + С02
Если раствор манганата оставите стоять на воздухе, то окраска его постепенно изменяется, переходя из зеленой в малиновую, причем образуется темно-бурый осадок. Это объясняется тем, что в водном растворе манганаты самопроизвольно превращаются в соли марганцовой кислоты НМn04 (перманганаты) с одновременным образованием диоксида марганца. Реакция выражается уравнением
ЗК2Мn04 + 2Н2О = 2КМn04 + Мn02 +4КОН.
манганат калия перманганат калия
или в ионно-молекулярной форме:
При этой реакции один ион Мn04 окисляет два других таких же иона в ионы Мn04 , а сам восстанавливается, образуя МnО2.
При действии сильных окислителей (например, хлора) па раствор манганата последний полностью превращается в перманганат:
2К2Мn04 + С12 = 2КМn04 + 2КС1.
Перманганат калия КМn04 — наиболее широко применяемая соль марганцовой кислоты. Кристаллизуется он в виде красивых темно-фиолетовых, почти черных призм, умеренно растворимых в воде. Растворы КМn04 имеют темно- малиновый, а при больших концентрациях — фиолетовый цвет, свойственный ионам МnО-. Как и все соединения марганца (VII), перманганат калия - сильный окислитель. Он легко окисляет многие органические вещества, превращает соли железа (II) в соли железа (III), сернистую кислоту окисляет в серную, из соляной кислоты выделяет хлор и т. д.
Вступая в окислительно-восстановительные реакции, КМn04 (ион Мn04) может восстанавливаться в различной степени. В зависимости от pH среды продукт восстановления может представлять собой ион Мn2+ (в кислой среде), МnO2 (в нейтральной или в слабо щелочной среде) или ион МпО42- (в сильно щелочной среде).
Проиллюстрируем эти три случая реакциями взаимодействия КМn04 с растворимыми сульфитами. Если к подкисленному серной кислотой фиолетовому раствору КМп04 прибавить сульфит калия K2S03, то жидкость становится почти бесцветной, так как образующаяся соль марганца (II) имеет бледно-розовую окраску. Реакция выражается уравнением
2КМn04 + 5K2S03 + 3H2S04 = 2MnS04 + 6K2S04 + 3H20
При действии сульфита калия на нейтральный раствор перманганата калия тоже происходит обесцвечивание раствора, но, кроме того, выпадает бурый осадок диоксида марганца и раствор приобретает шелочную реакцию
2КМn04 + 3K2S03 + Н20 = 2Мn02 +3K2S04 + 2КОН
При большой концентрации щелочи и малом количестве восстановителя образуются ионы манганата согласно уравнениям:
2КМn04 + K2S03 + 2КОН = 2K2Mn04 + K2S04 + Н20;
При нагревании в сухом виде перманганат калия уже при температуре около 200 °С разлагается согласно уравнению:
2КМn04 = К2Мn04 + Мn02 + 02
Оксид марганца (VII), или марганцовый ангидрид, Мn2O7 может быть получен действием концентрированной серной кислоты на перманганат калия:
2KMn04 + H2S04 = Мn207 + K2S04 + Н20.
Марганцовый ангидрид — зеленовато-бурая маслянистая жидкость. Он очень неустойчив: при нагревании или при соприкосновении с горючими веществами он со взрывом разлагается на диоксид марганца и кислород.
Билет 11
Химия хрома
 Внешняя электронная оболочка атомов элементов подгруппы хрома содержит один или два электрона, что обусловливает металлический характер этих элементов и их отличие от элементов главной подгруппы. Вместе с тем их максимальная степень окисления равна +6, так как, помимо наружных электронов, в образовании связей может участвовать еще соответствующее число электронов из недостроенной предпоследней оболочки.
Внешняя электронная оболочка атомов элементов подгруппы хрома содержит один или два электрона, что обусловливает металлический характер этих элементов и их отличие от элементов главной подгруппы. Вместе с тем их максимальная степень окисления равна +6, так как, помимо наружных электронов, в образовании связей может участвовать еще соответствующее число электронов из недостроенной предпоследней оболочки.
Для хрома и его аналогов наиболее типичны производные высшей степени окисления, во многом сходные с соответствующими соединениями серы. Соединения хрома (VI) отличаются неустойчивостью в растворах и являются сильными окислителями. При этом они чаще всего восстанавливаются до анионных или катионных комплексов хрома (III). Хотя хром располагается в четной группе, наиболее устойчивой его степенью окисления является +3. Это связано с тем, что соединения хрома (III) являются, как правило, комплексными с координационным числом 6 и октаэдрической пространственной конфигурацией расположения лигандов. В этом случае три Зй-электрона иона Сг3+ равномерно заселяют трижды вырожденные несвязываюшие МО комплекса. Возникающая стабилизация системы за счет суммарного спина 3 * l/2 = 3/2 (по правилу Хунда) в этом случае больше, чем если бы степень окисления хрома была +2, +4 и т. д.
Хром содержится в земной коре. В природе он встречается главным образом в виде хромистого железняка FeO*Cr2O3.
Хром представляет собой твердый блестящий металл. При комнатной температуре хром стоек и к воде, и к воздуху. Разбавленные серная и соляная кислоты растворяют хром с выделением водорода. В холодной концентрированной азотной кислоте хром нерастворим и после обработки ею становится пассивным.
Металлический хром используется для хромирования, а также в качестве одного из важнейших компонентов легированных сталей. Введение хрома в сталь повышает ее устойчивость против коррозии как в водных средах при обычных температурах, так и в газах при повышенных температурах.
Хром образует три оксида: оксид хрома (II) СгО, имеющий основной характер, оксид хрома (III) Сг203, проявляющий амфотерные свойства, и оксид хрома (VI), или хромовый ангидрид, СгО3 — кислотный оксид. Соответственно этим трем оксидам известны и три ряда соединений хрома.
Соединения хрома (II). При растворении хрома в соляной кислоте получается раствор голубого цвета, содержащий хлорид хрома (II) СгС12. Если к этому раствору прилить щелочи, то выпадает желтый осадок - гидроксид хрома (II) Сг(0Н)2. Соединения хрома (II) неустойчивы и быстро окисляются кислородом воздуха в соединения хрома (III).
Соединения хрома (III). Оксид хрома (III) Сг203 представляет собой вещество зеленого цвета.
Гидроксид хрома (III) Сг(ОН)3 выпадает в виде синевато-серого осадка при действии щелочей на соли хрома (III):
Сг3+ + 0Н- = Сг(ОН)3
Подобно гидроксидам алюминия и цинка, он имеет амфотерный характер и растворяется в кислотах с образованием солей хрома (III), а в щелочах — с образованием изумрудно-зеленых растворов гидроксохроматов (III) (иначе их называют хромитами), например:
Cr(OH)3 + 3NaOH = Na3[Cr(OH)6]
Хромиты, полученные сплавлением Сг203 с оксидами других металлов и известные главным образом для двухвалентных металлов, имеют состав, отвечающий формуле Ме(Сг02)2, и представляют собой соли метахромовой(III) кислоты (иначе — метахромистой кислоты) НСг02. К ним относится и природный хромистый железняк Fe(CrO2)2.
Из солей хрома (III) самой распространенной является двойная соль хрома и калия — хромокалиевые квасцы KCr(S04)2*12H20, образующие синефиолетовые кристаллы.
Соли хрома (III) во многом похожи на соли алюминия. В водных растворах они сильно гидролизованы и легко превращаются в основные соли. Со слабыми кислотами хром (III), подобно алюминию, солей не образует.
Растворы солей хрома (III) обычно имеют синефиолетовый цвет, но при нагревании становятся зелеными, а спустя некоторое время после охлаждения основа приобретают прежнюю окраску. Это изменение окраски объясняется образованием изомерных гидратов солей, представляющих собой комплексные соединения, в которых все или часть молекул воды координационно связаны во внутренней сфере комплекса. В некоторых случаях такие гидраты удалось выделить в твердом виде. Так, кристаллогидрат хлорида хрома (III) СгClз*6Н2О известен в трех изомерных формах: в виде сине-фиолетовых, темно-зеленых и светло-зеленых кристаллов одинакового состава. Строение этих изомеров можно установить на основании различного отношения их свежеприготовленных растворов к нитрату серебра. При действии последнего на раствор сине-фиолетового гидрата осаждается весь хлор; из раствора темно-зеленого гидрата осаждается 2/3 хлора, а из раствора светло-зеленого гидрата — только 1/3 хлора. Принимая во внимание эти данные, а также координационное число хрома, равное шести, строение рассматриваемых кристаллогидратов можно выразить следующими форму-лами:
[Сг(Н20)6]С13 [Сг(Н20)5С1]С12*Н20 [Сг(Н20)4С12]С1*2Н20
сине-фиолетовый темно-зеленый светло-зеленый
Таким образом, изомерия гидратов хлорида хрома (III) обусловлена различным распределением одних и тех же групп (Н20 и С1) между внутренней и внешней координационными сферами и может служить примером гидратной изомерии.
Соединения хрома (VI). Важнейшими соединениями хрома (VI) являются триоксид хрома, или хромовый ангидрид, СгОз и соли отвечающих ему кислот — хромовой Н2СrO4 и дихромовой Н2Сr2О7. Обе кислоты существуют только в водном растворе и при попытках выделить их из раствора распадаются на хромовый ангидрид и воду; но соли их достаточно стойки. Соли хромовой кислоты называются хромитами, а дихромовой - дихроматами.
Почти все хроматы имеют желтую окраску.
При подкислении раствора какого-нибудь хромата, например хромата калия К2СrO4, чисто-желтая окраска раствора сменяется на оранжевую вследствие перехода ионов СгО42- в ионы Сr2О72-. Из полученного раствора может быть выделена соль дихромовой кислоты — дихромат калия К2Сr2О7 — в виде оранжево-красных кристаллов. Реакция превращения хромата в дихромат выражается уравнением:
2СrO42- + 2Н+ Сr2072~ + Н20.
Реакция обратима. Это значит, что при растворении дихромата в воде всегда образуется некоторое, хотя и незначительное, количество ионов Н+ и СгО42-; поэтому раствор дихромата имеет кислую реакцию. Если к раствору дихромата прибавлять щелочь, то гидроксид-ионы будут связывать находящиеся и растворе ионы водорода, равновесие смещается влево и в результате дихромат превращается в хромат. Таким образом, в присутствии избытка гидроксид-ионов в растворе практически существуют только ионы СгО42-, т. е. хромат, а при избытке ионов водорода — ионы Сг2О7 2- , т. е. дихромат.
Хроматы щелочных металлов получаются путем окисления соединений хрома (III) в присутствии щелочи. Так, при действии брома на раствор хромита калия образуется хромат калия:
2К3[Сг(ОН)6] + ЗВг2 + 4К0Н = 2К2Сг04 + 6КВг + 8Н20.
О происходящем окислении можно судить по тому, что изумрудно-зеленая окраска раствора хромита переходит в ярко-желтую.
Хроматы могут быть получены также сплавлением Сr2О3 с щелочью в присутствии какого-нибудь окислителя, например хлората калия:
Сг203 + 4К0Н + КСlO3 = 2К2Сг04 + КС1 + 2Н20 .
Хроматы и дихроматы — сильные окислители. Поэтому ими широко пользуются для окисления различных веществ. Окисление проводится в кислом растворе и обычно сопровождается резким изменением окраски (дихроматы окрашены в оранжевый цвет, а соли хрома (III) — в зеленый или зеленовато-фиолетовый).
Мы видели, что в кислых и в щелочных растворах соединения хрома (III) и хрома (VI) существуют в разных формах: в кислой среде в виде ионов Сг3+ или Сr2О72- , а в щелочной — в виде ионов [Сг(ОН)6]3 или СгО42-. Поэтому взаимопревращение соединений хрома (III) и хрома (VI) протекает по-разному в зависимости от реакции раствора. В кислой среде устанавливается равновесие
Сг2072- + 14Н+ + 6е 2Сг3+ +
Дата добавления: 2016-10-07; просмотров: 4508;
Поиск по сайту
Узнать еще
- АТРИОВЕНТРИКУЛЯРНАЯ БЛОКАДА ВТОРОЙ СТЕПЕНИ
- АТРИОВЕНТРИКУЛЯРНАЯ БЛОКАДА ВЫСОКОЙ СТЕПЕНИ
- В зависимости от степени точности
- Ведущие факторы в оценке степени тяжести гестоза
- Влияние степени переохлаждения на процесс кристаллизации
- Выбор электрооборудования по степени защиты
- Высокая объемная эффективность (VE) приводит к высокой динамической степени сжатия.
- ГОСТ 28245-89 Торф. Методы определения ботанического состава и степени разложения

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
