СТРУКТУРА И ФУНКЦИИ ИММУННОЙ СИСТЕМЫ 15 глава
Известно два способа смерти живой клетки — некроз и апоптоз.
Некроз — травматическая гибель по самым разным причинам (механическое или осмотическое нарушение целостности мембраны, ожог термический или химический, повреждение микробными или иными токсинами и т.д.). Некроз приводит к попаданию содержимого клетки в окружающую среду и в норме вызывает воспалительную реакцию.
Апоптоз — фрагментация содержимого клетки изнутри, осуществляемая специальными внутриклеточными ферментами, индукция и активация которых происходит при получении клеткой внешнего сигнала или при принудительной «инъекции» в клетку ферментов–активаторов апоптозной «машины» (последнее имеет место при осуществлении эффекторной функции ЦТЛ при убийстве ими клеток–мишеней). Апоптоз развивается и при повреждении клетки внешними факторами, не приводящими к некрозу, но способными инициировать апоптоз (ионизирующая радиация, обратимый перегрев и др.).
Явление одновременной гибели определённых клеток в многоклеточном организме наблюдали ещё в конце прошлого века — начале нашего и именно на лимфоцитах. Физиологи регистрировали, что при голодании в первую очередь организм теряет значительную часть лимфоцитов. Поскольку функции лимфоцитов в те времена были неизвестны, их даже называли трофоцитами как раз за то, что организм «съедает» их в первую очередь при голодании. Наблюдали также безвоспалительную гибель тимоцитов при введении в организм экспериментальных мышей глюкокортикоидных гормонов. Термин «апоптоз» тогда ещё не применяли.
Апоптозом более 20 лет назад назвали биологическое явление, описанное при гистологическом исследовании развития личинок круглых червей — почвенной нематоды Caenorhabditis elegans. У червей наблюдали одновременную гибель одного и того же числа клеток в строго определённый момент развития: тело личинки содержит 1076 клеток, а тело взрослой нематоды — 945, и ровно 131 клетка в нужный момент исчезает. Эта «ровность» навела на правильную мысль о запрограммированности такой клеточной смерти.
Термин «апоптоз» происходит от греческого apoptosis — листопад. У нематод идентифицировали два гена — ced–3 и ced–4, необходимые для осуществления апоптоза, и один ген ced–9, подавляющий апоптоз. В 1976 г. были опубликованы результаты биохимических экспериментов, в которых при электрофорезе ДНК обнаружили факт фрагментации ДНК на регулярные отрезки размером примерно 180 пар нуклеотидов. Как всякая регулярность и правильность, а не хаос это также навело на верную мысль о том, что в данном случае учёные столкнулись с закономерным явлением природы, имеющим отлаженный биохимический и клеточный механизм развития. Но с феноменом запрограммированной гибели клеток этот факт был «увязан» позже. У млекопитающих клетки всякой ткани, которой свойственна пожизненная физиологическая регенерация, в дифференцировке устанавливают программу на апоптоз.
Примеры морфогенетических событий, при которых происходит апоптоз, как минимум следующие:
· удаление стадиоспецифичных тканей и органов в эмбриогенезе;
· удаление короткоживущих (несколько часов) клеток крови (например, нейтрофилов);
· позитивная и негативная селекция тимоцитов;
· элиминация пролиферирующих клеток в отсутствие их специфических факторов роста;
· индуцированная активацией смерть лимфоцитов.
Но вообще говоря, во всякой живой ткани есть какое-то количество клеток, претерпевающих апоптоз, ибо все ткани с той или иной скоростью обновляются, пока организм жив. Кроме того, апоптоз запускается в клетках–мишенях при работе таких эффекторов иммунитета, как ЦТЛ и NK, в том числе как эффекторы антителозависимой клеточной цитотоксичности.
У периферических лимфоцитов есть три Рц и три сигнала на апоптоз — Fas–FasL, TNFR1–TNF, Рц для глюкокортикоидов–глюкокортикоиды. Экспрессия этих Рц инициируется вместе с началом иммунного ответа и нарастает по мере активации и дифференцировки иммунных лимфоцитов. Всего же в иммунной системе не только в периферических тканях, но и при лимфопоэзе известны следующие пары Рц—лиганд, связывание которых индуцирует апоптоз в клетке — носителе Рц:
· Рц Fas (CD95) на клетке–мишени — Fas–лиганд (FasL) на мембране клетки–"убийцы», Рц Fas относится к семейству Рц для TNF;
· Рц для глюкокортикоидных гормонов в лимфоците — глюкокортикоидные гормоны. Образование этого комплекса как механизма индукции апоптоза «работает» на территории тимуса при позитивной селекции, причём глюкокортикоиды синтезируются эпителиальными клетками тимуса. На территории тимуса при негативной селекции тимоцитов апоптоз индуцирует Fas–лиганд, экспрессированный на дендритных клетках тимуса. Вероятно, глюкокортикоиды индуцируют апоптоз и активированных лимфоцитов в периферических тканях;
· при определённых сопутствующих условиях Рц для фактора некроза опухоли — TNF типа 1 (TNFR–1, p55) и его многочисленные лиганды (например, TNFa). В настоящее время выделяют до 6 так называемых Рц TRAIL (TNF–receptor–related apoptosis inducing ligand), родственных Рц для TNF, лиганды которых могут вызывать апоптоз;
· CD30 (на тимоцитах или T–лимфоцитах) — CD30L (на эпителии и дендритные клетки мозговой зоны тимуса и других клетках).
После связывания с лигандом Рц Fas тримеризуются. В цитоплазматических участках Рц Fas и TNFR–1 есть особые «домены смерти», обеспечивающие белок–белковое взаимодействие с адапторными белками FADD (связывается с Fas–Рц изнутри клетки) и TRADD (связывается с TNFR–1). Эти адапторные белки связывают особый фермент — цистеиновую протеазу каспазу–8. C–концевой участок этой молекулы является прокаспазой. Каким-то образом в начало апоптозного процесса вовлекаются митохондрии: повышается проницаемость их мембраны и из митохондрий в цитозоль высвобождается цитохром С. В цитозоле цитохром С образует комплекс с белком Apaf–1. Этот комплекс активирует каспазы, в первую очередь каспазу–8. Каспаза–8 запускает каскад активации других каспаз, последняя из которых (возможно, каспаза–3) активирует особую ДНКазу CAD. Эта ДНКаза разрезает ДНК клетки между нуклеосомами на куски размером примерно по 200 (50–300) пар нуклеотидов.
Каспазы — (относящиеся к апоптозу цистеиновые протеазы CASP) аспартатспецифические протеазы, ключевые ферменты воспаления и апоптоза, осуществляют деградацию множества клеточных белков, функционируют во внутриклеточных сигнальных путях на промежуточных и завершающих стадиях реализации апоптоза. В апоптозе участвуют два класса каспаз — инициаторы и эффекторы. Проапоптозным сигналом активируются инициаторные каспазы ‑2, –8, –9). Инициаторные каспазы процессируют эффекторные каспазы ‑3, –6, –7), действие которых и приводит к гибели клетки вследствие расщепления специфических субстратов, в первую очередь ферментов метаболизма нуклеиновых кислот (например, поли[АДФ-рибоза] полимераза, КФ 2.4.2.30).
Номенклатура каспаз. У человека известно 11 каспаз семейства ICE/CED–3 (ICE — от Interleukin 1b Converting Enzyme [каспаза 1], КФ 3.4.22.36; CED–3 — от CEll Death gene ced–3 почвенной нематоды Caenorhabditis elegans). Для членов этого семейства предложено тривиальное имя «каспаза» — «caspase» («c» от «cysteine»), «aspase» от «asp» («aspartic acid» — расщепление полипептида после остатка аспарагиновой кислоты). Для обозначения отдельных каспаз добавляется номер фермента (каспаза 1, каспаза 2, ..., каспаза 13).
Параллельно и, по-видимому, независимо от фрагментации ДНК наступает фрагментация остального содержимого клетки (рушится цитоскелет), и клетка сначала сморщивается и уменьшается в размерах, затем распадается на апоптозные пузырьки, или тельца. При этом внутриклеточное содержимое почти не вытекает во внеклеточную среду и в окружающей ткани не развивается воспаление. Поэтому тимус, в котором больше 95% тимоцитов, непрерывно погибает апоптозом, не болит, не отекает, не краснеет, не повышается его температура.
Каспазы связаны с кислыми и нейтральными сфингомиелиназами, которые, будучи активированными, генерируют церамиды и ганглиозиды, меняющие мембраны клетки. В результате ядро постепенно сморщивается и фрагментируется. Клеточная мембрана теряет упругость, местами происходит инверсия фосфолипидов и наружу «выворачивается» фосфатидилсерин. Его способны распознавать макрофаги и дендритные клетки (ДК). Затем клетка распадается на окружённые мембраной фрагменты — апоптозные тельца. Внутриклеточное содержимое при этом не «вытекает» во внеклеточную среду. Воспалительная реакция в ткани не развивается. Апоптозные тельца — мембранные пузырьки — фагоцитируют макрофаги либо поглощают ДК макроэндоцитозом. Макрофаги сорбируют апоптозные тельца следующими молекулами своей мембраны: интегрином avb3 молекулой CD36 (молекула адгезии, GPIIIb), Рц для «мусора» (scavenger receptor). ДК сорбируют апоптозные тельца интегрином avb3 и молекулой CD36.
Макрофаги разрушают вещество апоптозных телец до мелких метаболитов. При этом не активируется продукция провоспалительных цитокинов в макрофагах. А вот дендритные клетки перерабатывают вещества апоптозных телец, как и любых других Аг, и в результате экспрессируют на мембране комплексы пептидов с молекулами MHC–I и II. Среди пептидов находятся как экзогенные, например, вирусные (если апоптозу подверглась инфицированная вирусом клетка), так и пептиды — фрагменты собственных белков организма. Дендритные клетки способны индуцировать иммунный ответ на компоненты апоптозных телец. Именно такие процессы лежат в основе развития иммунного ответа на собственные повреждённые ткани, а также на тканевые Аг чужеродных трансплантатов.
Известны гены (по крайней мере два), продукты которых предотвращают апоптоз. Это гены Bcl–2 (соответственно белок bcl–2) и Bcl–x. Ген Bcl–2 млекопитающих гомологичен гену ced–9 у нематод. С гена Bcl–x в результате альтернативного сплайсинга первичного РНК–транскрипта получаются два белка — малый (small) bcl-xS и большой (large) bclL. Белок bcl–xS индуцирует апоптоз, а белок bclL апоптоз предотвращает. Механизм антиапоптозного действия белка bcl–2 предположительно известен: этот белок связывается с мембраной митохондрий и препятствует высвобождению из этих органелл цитохрома С, без чего апоптоз не начинается. Излишняя экспрессия этих антиапоптозных белков происходит при лимфопролиферативных заболеваниях и хронических воспалительных процессах, например при ревматоидном артрите в T–лимфоцитах синовиальных полостей. В последнем случае полагают, что антиапоптозные сигналы лимфоциты синовиальных полостей получают от разросшихся фибробластов стромы, что в патогенезе заболевания является первичным процессом по отношению к патогенетическим эффектам активированных T–лимфоцитов.
Сигналы для экспрессии Bcl–2 и Bcl–xL, вероятно, приходят с g–цепи Рц для цитокинов, общей у Рц для интерлейкинов 2, 15, 4, 7 и 13. Кроме того, при анализе механизмов хронического воспаления суставов при ревматоидном артрите установили, что индукция антиапоптозных генов в зрелых T–лимфоцитах (которым в норме положено было бы умереть апоптозом), вероятно, происходит при взаимодействии молекул интегринов на фибробластах (активный пептид Arg–Gly–Asp–Ser) с молекулой LFA–1 (CD11a/CD18) на T–лимфоцитах.
Кстати, в экспериментах с B–лимфоцитами показано, что активация B–лимфоцита через специфический Рц для Аг (BCR) сопряжена с защитой от апоптоза на несколько дней, необходимых для развития иммунного ответа, тогда как стимуляция B–лимфоцита, помимо BCR, например митогенами, приводит к их быстрому, опосредованному через Fas–Рц апоптозу.
Индукция так называемой высокодозовой толерантности (когда в организм парентерально вводят большую дозу специфического Аг) по механизму — это элиминация Аг–специфичного клона лимфоцитов апоптозом как следствие «ударной» активации клона специфическим Аг. Превышение меры — всегда патология.
В норме факторы транскрипции, образующиеся при нормальном ходе активации T–лимфоцита, — АP–1, NFAT, NFkB — активируют и энхансерные последовательности для Fas–лиганда, что создаёт перспективу для апоптоза лимфоцитам, «отработавшим» в иммунном ответе.
Суперантигены индуцируют апоптоз сразу после пролиферации поликлонально «охваченных» T–лимфоцитов, они также индуцируют апоптоз дендритных клеток. Все перечисленное объясняет продолжительную иммунодепрессию после воздействия на организм суперантигенов.
Описаны наследственные дефекты в Fas или/и FasL: такие люди с самого рождения страдают тяжёлыми формами лимфо-пролиферативных или/и аутоиммунных болезней, быстро приводящих к смерти.
У млекопитающих выявлены и другие гены, кодирующие продукты подавления апоптоза: Bcl–w, Mcl–1, ALG–3 и др. Гены, ингибирующие апоптоз «наших» клеток, найдены у вирусов Эпштейна–Барр, аденовирусов, вируса африканской лихорадки свиней. Наличие таких генов у вирусов положительно коррелирует с их онкогенностью. Поэтому не напрасно клиницисты рассматривают эти вирусы как потенциально онкогенные. Есть и гены, ускоряющие апоптоз. У млекопитающих выявлены такие гены —Bax и Bad. Белки, кодируемые этими генами, функционируют в виде димеров. Возможно образование димеров из проапоптозного белка bах с антиапоптозным белком bcl–2 (bax—bcl–2). От баланса про- и антиапоптозных белков в клетке зависит, будет лимфоцит жить или погибнет.
Таким образом, физиологический апоптоз сопряжен с активацией и пролиферацией лимфоцитов. Это сопряжение настолько обязательно, что феномен гибели лимфоцитов после активации получил отдельное название — «индуцированная активацией смерть клеток».
Однако апоптоз может быть инициирован в клетке и факторами физического проникающего патогенного внешнего воздействия. Например, апоптоз запускается в клетках, в которых произошло радиационное повреждение ДНК в большем объёме, чем могут «исправить» ферменты репарации ДНК. Некроза клеток при этом нет, есть именно апоптоз. Внутриклеточным фактором, инициирующим программу на апоптоз в данном случае, вероятно, является некий белок с молекулярной массой 53 кДа, поэтому он обозначен p53. Физиологические функции этого белка неизвестны, неизвестно даже, функционирует ли он в норме. Но вот при дефектах в гене p53 развиваются опухоли. У нокаутированных мышей по гену p53 онтогенез примерно до 10–месячного возраста проходит без видимых отклонений, но после 10 мес практически все такие животные умирают от рака или саркомы. Так или иначе к настоящему времени сложилось представление, что белок p53 необходим для инициации апоптоза в клетках, в которых повреждена ДНК. Причины повреждений ДНК могут быть самые разные: вирусные инфекции, ионизирующая радиация, химический мутагенез и др. Если клетка не справляется с репарацией наступивших повреждений ДНК, то p53 запускает в этой клетке апоптоз. Эта рабочая гипотеза достаточно эффективно оправдывает себя в прикладных исследованиях по поиску фармакологических средств, предназначенных для защиты клеток от апоптоза. Такая проблема существует у онкологических больных во время курсов интенсивной противоопухолевой химиотерапии. Современные химиотерапевтические препараты — это цитостатики, большинство из них — антиметаболиты ДНК. Они токсичны не только для клеток опухоли, но и других делящихся клеток — эпителия слизистых оболочек и кожи, всех ростков кроветворения и др. Поэтому у больных в качестве побочных эффектов лечения развиваются анемия, лейкопения, тромбоцитопения, выпадают волосы, возникают проблемы с работой кишечника. Появилась и успешно разрабатывается идея временного фармакологического подавления в организме p53 только на период курса противоопухолевой химиотерапии. Временное отсутствие p53 спасет разные клетки от апоптоза, потенциально вызываемого химиопрепаратами, кроме клеток самой опухоли, поскольку в ней p53 и так не функционирует. Или, например, если тимоциты in vitro поместить в среду с температурой 43 °C, то они через некоторое время погибнут по механизму некроза. Но если те же тимоциты прогреть до той же температуры 43 °C в течение 3 мин, после чего «вернуть» к физиологической температуре 36,7 °C, то тимоциты будут все равно погибать, но по механизму апоптоза. Кроме того, постнатальный апоптоз возможен и не в лимфоидной ткани, например апоптоз гепатоцитов при алкогольных циррозах печени, при которых обнаружили экспрессию Fas и FasL именно на гепатоцитах.
· Глава 7. ИММУННЫЙ ОТВЕТ
7.1. Определение иммунного ответа. Этапы иммунного ответа
определили иммунитет как особое биологическое свойство и показали его место среди других биологических механизмов защиты многоклеточных организмов от инфекций. Иммунный ответ — это иммунитет в действии, т.е. процесс иммунного воспаления, инициированный патогеном. Кратко определить иммунный ответ можно ещё и так:
Распознавание повреждённых патогеном клеток и тканей организма лимфоцитами с целью деструкции и выведения из организма.
Элиминация из организма тканей, повреждённых патогеном, в ходе воспалительного процесса с участием лимфоцитов.
Доиммунное воспаление + распознавание повреждённых клеток лимфоцитами + деструкция повреждённых тканей в ходе иммунного воспалительного процесса + выведение продуктов деструкции из организма.
Дадим ещё раз в конспективном виде характеристики иммунного ответа, по которым его можно отличить от других защитных процессов, с одной стороны, и увидеть его сопряженность с другими защитными механизмами — с другой. Это и будет длинное, но конкретное определение иммунного ответа: иммунный ответ — это такой процесс, в котором можно найти совокупность перечисленных ниже характеристик:
1) иммунный ответ — воспалительный процесс с обязательным участием лимфоцитов;
2) лимфоциты — единственный тип клеток в организме, в дифференцировке которых происходит обязательная соматическая рекомбинация ДНК и именно сегментов генов Рц для Аг. В результате в течение всего онтогенеза в организме непрерывно генерируется беспрецедентное разнообразие клонов лимфоцитов по Рц для Аг — 1018 вариантов у T–лимфоцитов и 1016 вариантов у B–лимфоцитов;
3) кроме Рц для Аг, на мембране лимфоцитов есть инвариантные, но строго необходимые для развития иммунного ответа корецепторы;
4) связывание Рц лимфоцита для Аг своего лиганда необходимо, но недостаточно для инициации иммунного ответа;
5) Рц T–лимфоцита для Аг распознаёт (связывает) повреждённые (изменённые) патогеном структуры собственных тканей, а не патоген сам по себе как таковой;
6) для инициации иммунного ответа лимфоциту необходимо получить сигналы по «двум каналам» — с Рц для Аг и с корецепторов;
7) сигналы с корецепторов поступают к лимфоцитам от клеток доиммунного воспаления (дендритных клеток, макрофагов, покровных эпителиев) и их биологически активных продуктов;
8) клетки доиммунного воспаления не имеют большого разнообразия Рц, их Рц инвариантны, консервативны, кодируются зародышевыми генами, но именно эти Рц первыми избирательно связывают продукты микроорганизмов, не характерные для макроорганизмов. Следовательно, именно Рц клеток доиммунного воспаления — «носители эволюционной памяти», которые первыми отличают «чужое» от «своего» и биохимически информируют лимфоциты о факте проникновения чужого во внутреннюю среду;
9) Рц лимфоцита для Аг направляет иммунные лимфоциты (как вектор) к своим тканям, повреждённым патогеном;
10) лимфоцит при инициации иммунного ответа связывает свой лиганд в лимфоидных органах, активируется, обязательно пролиферирует, додифференцируется и мигрирует в кровь. В очаге поражения уже иммунный лимфоцит выходит из крови в ткань, там вновь связывает Рц для Аг свой лиганд и либо сам подвергает повреждённые клетки деструкции (ЦТЛ), либо выделяет цитокины, «нанимающие» на деструкцию макрофаги, эозинофилы, нормальные киллеры и другие лейкоциты (рис. 7.1).
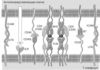
Рис. 7.1. Иммунологический синапс между T–лимфоцитом и АПК. Минимальный набор взаимодействий, необходимых для начала развития иммунного ответа: 1 — связь агрегированных TCR с комплексами пептид—MHC; 2 — связь корецептора CD4 (или CD8) с MHC; 3 — связь CD40 с CD40L; 4 — связь B7 с CD28; 5 — связи молекул адгезии (ICAM, LFA); 6 — взаимодействия цитокинов с Рц для цитокинов.
Иммунное воспаление — воспаление с участием лимфоцитов. Иммунное воспаление — наиболее активное и агрессивное по сравнению с доиммунным воспалением, т.е. вариантами воспалительных процессов, развивающихся без вовлечения в них лимфоцитов.
Этапы развития иммунного ответа во времени таковы:
· доиммунное воспаление в очаге внедрения патогена; распознавание и поглощение патогенов и их продуктов дендритными клетками (АПК);
· представление Аг лимфоцитам в лимфоидных органах;
· пролиферация лимфоцитов;
· эффекторная дифференцировка лимфоцитов;
· иммунное воспаление с деструкцией тканей, повреждённых патогеном;
· регенерация тканей в очаге повреждения.
Более подробно можно описать следующие события и процессы.
· Всё начинается с проникновения Аг во внутреннюю среду организма. В природе это происходит при травмировании покровных тканей. При этом в покровных тканях выделяются определённые вещества (стресc–протеины, белки теплового шока, цитокины кератиноцитов и клеток соединительной ткани) — медиаторы доиммунного воспаления, которые готовят почву для развития, если понадобится, лимфоцитарного имммунного воспаления. Попадание Аг без значимой травмы покровов сразу во внутреннюю среду в природе — событие редкое. Это, как правило, искусственное антропогенное явление парентерального введения веществ или трансплантации тканей и органов.
· Доиммунные защитные реакции в отношении Аг направлены на то, чтобы не пустить Аг глубже покровов. В первую очередь это сосудистые реакции: расширение сосудов микроциркуляторного русла, повышенный выпот из сосудов в ткани плазмы или сыворотки (соответственно всех сывороточных факторов доиммунной резистентности к инфекциям) и экстравазация лейкоцитов, в первую очередь фагоцитов — нейтрофилов. Локальный отёк препятствует всасыванию Аг в системную циркуляцию.
à Проникший в покровы патоген сорбируют и поглощают эндоцитозом дендритные клетки, фагоцитируют макрофаги. И те и другие — профессиональные АПК, но только дендритные клетки обладают особыми способностями — мигрируют из покровов с Аг в регионарные лимфоидные органы. «По дороге» дендритные клетки процессируют Аг, экспрессируют на мембрану комплексы пептидов с MHC–I и MHC–II и необходимые корецепторные молекулы, с помощью которых они смогут вступить в эффективное воздействие с T–лимфоцитами в T–зависимых зонах периферических лимфоидных органов. Кроме АПК, в покровных тканях Аг встречают внутриэпителиальные лимфоциты, среди которых много Tgd, распознающих непептидные Аг без предварительного процессинга и презентации АПК. Под покровами, в плевральной и брюшной полостях, для перехвата широко распространённых микробных Аг существуют АТ с широкой перекрестной реактивностью — продукты B1–лимфоцитов.
à «Неперехваченный» в барьерных тканях Аг, всосавшийся в системную циркуляцию, если он опасен для организма, сразу начнет приносить вред. Шанс на развитие иммунного ответа на него обусловлен тем, что есть такой лимфоидный орган, как селезёнка, через которую проходит весь объём крови за цикл циркуляции, и АПК в синусоидах селезёнки (дендритные клетки и макрофаги) будут пытаться сорбировать Аг.
· Пришедшие в лимфатические узлы дендритные клетки с Аг (их называют «интердигитирующие дендритные клетки» в отличие от иных, фолликулярных, дендритных клеток) располагаются в T–зависимых зонах и представляют Аг для «рассмотрения» интенсивно мигрирующим T–лимфоцитам. Среди T–лимфоцитов рано или поздно найдется такая клетка, у которой Рц для Аг окажется комплементарным данному Аг (т.е. свяжет Аг/MHC с константной диссоциации по абсолютной величине показателя степени не ниже чем 10–6–10–9 М). Если при этом состоятся все необходимые и достаточные корецепторные взаимодействия с АПК (см. рис. 7.1), то T–лимфоцит получит активационный сигнал, и с этого момента начнется собственно иммунный ответ — лимфоцитарный.
· T–лимфоцит начнет пролиферироватъ и дифференцироваться. В результате образуется клон антигенспецифичных дифференцированных T–лимфоцитов. Такие T–лимфоциты уже называют иммунными лимфоцитами, лимфоцитами-эффекторами. В процессе дифференцировки T–лимфоцит экспрессирует в надлежащем количестве мембранные молекулы и цитокины, необходимые для взаимодействия с B–лимфоцитами, лейкоцитами или для атаки на клетки–мишени.
· В T–зависимых зонах периферических лимфоидных органов происходит взаимодействие активированных Аг T–лимфоцитов с активированными Аг B–лимфоцитами.
· Провзаимодействовавший с Аг и с T–лимфоцитами B–лимфоцит мигрирует в зону фолликула, где пролиферирует и дифференцируется в антителопродуцента — плазматическую клетку. Сюда же, на территорию фолликула, мигрируют и Тh2–лимфоциты, на которых в результате взаимодействия с активированными Аг B–лимфоцитами экспрессируется особый Рц к хемокину — blr–1, обеспечивающий избирательное движение на территорию фолликула. Первые плазматические клетки остаются в лимфатическом узле, и секретируемые ими АТ в значительном количестве остаются здесь же — на Fc–Рц фолликулярных дендритных клеток (ФДК). V–области этих АТ связывают свой Аг. В таком виде в комплексе с АТ, фиксированными на ФДК, Аг может оставаться на территории лимфоидного фолликула в течение продолжительного времени (может быть месяцы и годы). Здесь, в фолликулах, при взаимодействии с этим Аг пойдет процесс созревания аффинности АТ, т.е. соматического гипермутагенеза генов V–бласти иммуноглобулинов и отбора (положительный сигнал на выживание) B–лимфоцитов с наиболее высокоаффинными вариантами АТ — только они и будут выживать и пролиферировать при вторичном иммунном ответе.
· Иммунные B–лимфоциты, дифференцировавшиеся в плазмоциты, уходят из фолликулов лимфоидных органов и мигрируют преимущественно в костный мозг или слизистые оболочки, где и «отрабатывают» массовую продукцию секретируемых в кровь или в слизистые секреты АТ. Плазмоциты из B–лимфоцитов лимфоидной ткани слизистых оболочек, продуцирующие АТ класса А (а также, вероятно, в определённой мере и Е), предназначенные для экскреции в слизистые экзосекреты, остаются для массовой продукции иммуноглобулинов в слизистой оболочке.
· Иммунные T–лимфоциты-эффекторы (ЦТЛ, Тh1, Th2) выходят из регионарных лимфатических узлов через эфферентные лимфатические сосуды, попадают в грудной лимфатический проток и оттуда в системную циркуляцию. Дальше они мигрируют через кровь в очаг воспаления в месте проникновения или диссеминации патогена: молекулы адгезии на иммунном лимфоците иные, чем на неиммунном. Иммунный лимфоцит «узнает» эндотелий сосудов микроциркуляции именно в очагах повреждения тканей и воспаления. Там TCR, если находит, то связывает свой Аг, что активирует лимфоцит. Состояние активации в данном случае заключается в усиленном биосинтезе и секреции эффекторных молекул. В случае ЦТЛ это молекулы, обеспечивающие убийство клеток–мишеней, в случае CD4+ Th — это цитокины, «нанимающие» для деструкции Аг те или иные лейкоциты (макрофаги, эозинофилы, тучные клетки, базофилы, нейтрофилы).
· Связанный Аг подвергается фагоцитозу и разрушению гидролитическими ферментами, кислородными радикалами, радикалами окиси азота до мелких метаболитов, которые экскретируются из организма через системы выделения (почки, ЖКТ).
· Организм санирован — первый результат достигнут.
· Супрессия иммунного ответа — остановка продуктивного иммунного ответа после санации организма от патогена/Аг.
· Второй результат лимфоцитарной иммунной реакции — иммунологическая память. По современным представлениям, лимфоцитами иммунологической памяти становится некая часть иммунных лимфоцитов (вероятно, порядка единиц процентов), на которых экспрессировались особые ингибирующие активацию Рц.
7.2. Иммунологическая память
Доиммунные механизмы резистентности «не запоминают» свою реакцию на Аг (патоген): он попадает в организм первый раз или десятый — реакция будет одинаковой (при одинаковом общем состоянии здоровья организма). Лимфоцитарный иммунитет «запоминает». Феномен иммунологической памятипроявляется в том, что в случае успешного иммунного ответа при первом попадании патогена в организм, при его повторных попаданиях санация наступает существенно быстрее и эффективнее, и патоген не успевает вызвать патологический инфекционный процесс. Это и называют протективным, т.е. защищающим от болезни иммунитетом.
В основе феномена иммунологической памятилежат два явления:
Дата добавления: 2016-07-18; просмотров: 1927;
Поиск по сайту
Узнать еще
- a-спираль b-складчатая структура
- Cимпатическая нервная система. Центральный и периферический отдел симпатической нервной системы.
- I. Общие принципы структурно-функциональной организации клетки и её компоненты. Плазмолемма, её структура и функции.
- I.2. Антигены системы АВ0. Генетика. Структура
- I.2.1 ПЕРВЫЙ ЗАКОН НЬЮТОНА. ИНЕРЦИАЛЬНЫЕ СИСТЕМЫ ОТСЧЁТА.
- I.2.1 ПОЛНАЯ И ВНУТРЕННЯЯ ЭНЕРГИЯ СИСТЕМЫ. ТЕПЛОТА И РАБОТА
- I.3. Антитела системы АВ0
- I.5.4 НЕИНЕРЦИАЛЬНЫЕ СИСТЕМЫ ОТСЧЁТА. СИЛЫ ИНЕРЦИИ

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
