Новый энергетический параметр стабильности кристаллического вещества - энергия сцепления атомных остовов и связующих электронов
До недавнего времени были известны два энергетических подхода к кристаллическому веществу: 1) основанный на концепции энергии кристаллической ионной решетки (энергии разрыва кристалла на свободные составляющие ионы) и 2) основанный на концепции энергии атомизации (энергии разрыва кристалла на свободные нейтральные составляющие атомы).
Следует признать, что по сравнению с ведущим вторым подходом первый энергетический подход имеет менее универсальный характер и соответственно ограниченные возможности в смысле предсказания физических свойств разнообразных кристаллических соединений и, в частности, металлов. Поэтому некоторые современные ученые не только предают забвению первый энергетический подход, но и фактически предлагают от него отказаться, как от устаревшего и неудовлетворительного. Хотя мы и не вполне разделяем эту точку зрения, однако в указанной ситуации разработка (предложение) еще одного принципиально нового конкурентноспособного энергетического подхода к кристаллам (и оценке их разнообразных физических свойств) представляется вполне оправданной, актуальной и целесообразной.
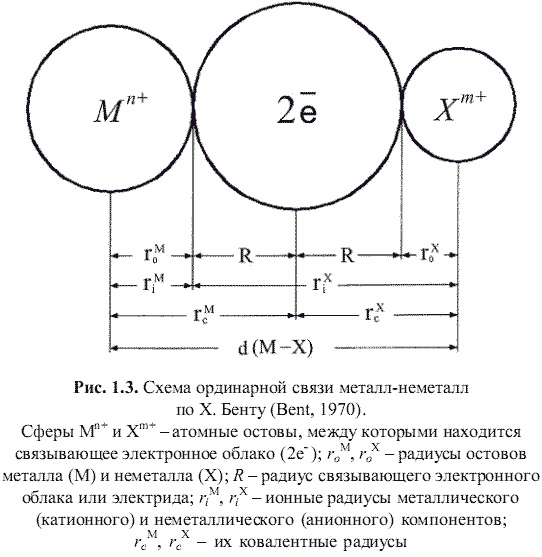
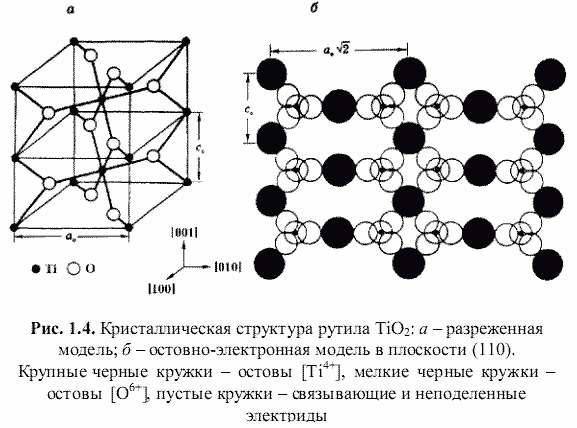
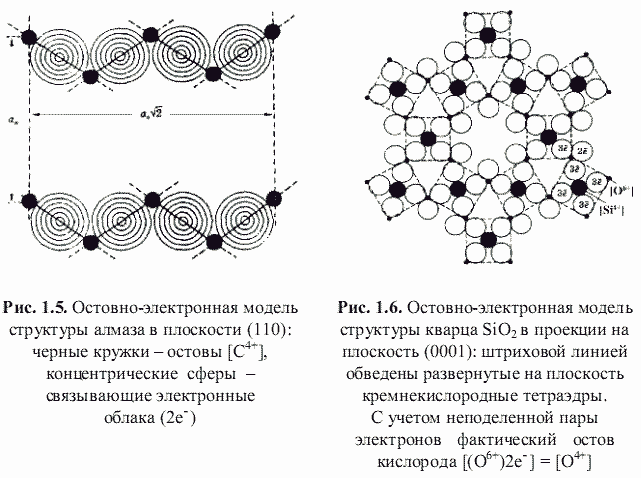
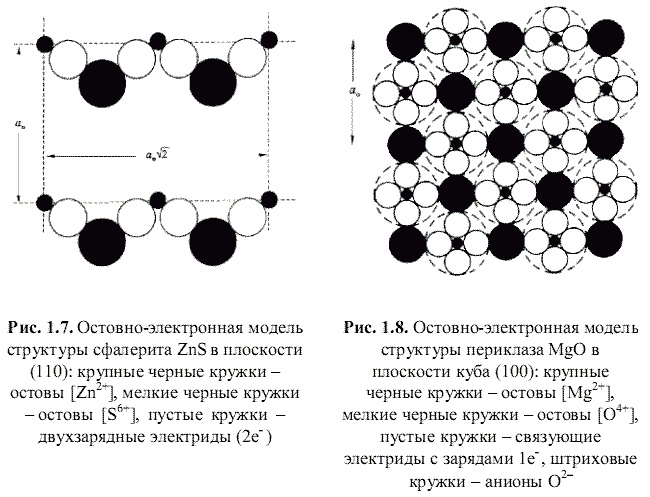
Как оказалось, теоретически возможен также третий энергетический подход к кристаллам, основанный на остовно-электронном моделировании их строения (Зуев, 1990). Напомним, что остовно-электронная концепция рассматривает кристаллическое соединение, независимо от типа химической связи, состоящим из положительно заряженных атомных остовов[1] (как металла, так и неметалла) и связывающих или поделенных (а также несвязывающих или неподеленных) валентных электронов. Принимая для тех и других, согласно (Bent, 1970; Зуев, 1990), сферическую форму (рис. 1.3), были построены соответствующие остовно-электронные модели кристаллических структур минералов, примеры которых приведены на рис. 1.4-1.8. Связывающие электронные облака можно именовать электридами, а соответствующие модели - остовно-электридными. Таким путем удается описать (хотя и с известной долей упрощения) строение соединений всех без исключения классов - будь то металлы, ковалентные или ионные кристаллы.[2]
Естественно предположить, что основу сцепления положительных остовов и отрицательных электридов в моделях типа изображенных на рис. 1.4-1.8 будет составлять электростатическое взаимодействие, энергию которого можно рассчитать в рамках концепции кристаллической ионной решетки, в которой атомные остовы являются катионами, а электриды - анионами. И, как нам представляется, наряду с энергией классической ионной решетки и энергией атомизации (используемых в первом и втором энергетических подходах), энергия остовно-электронного взаимодействия кристаллов в рамках третьего энергетического подхода может служить объективной мерой их стабильности и использоваться для оценки их свойств подобно тому, как это показано в работах (Зуев, Мочалов, Щербатов, 1998; Зуев, Аксенова, Мочалов и др., 1999). Схематически различие трех указанных энергетических подходов лучше всего продемонстрировать на конкретном примере обладающего тетраэдрической координацией атомов кристалла ZnS (сфалерита):
1. ZnS → Zn2+ + S2- + U;
2. ZnS → Zn0 + S0 + Eа;
3. ZnS → Zn2+ + S6+ + 8e- + W,
где выражаемые в кДж/моль или МДж/моль соответственно: U - энергия кристаллической решетки, Еа - энергия атомизации (энергия сцепления атомов), W - энергия сцепления двух остовов (Zn2+, S6+) и четырех двухзарядных электридов (4x2e-).
Соответствующая остовно-электронная модель для сфалерита, согласно рис. 1.7, -|[Zn2+](2e-)4[S6+]| т. е. на формульную единицу сфалерита приходится шесть заряженных частиц (два остова и четыре электрида). Остовно-электронная модель периклаза (рис. 1.8) - |[Mg2+](1e-)6[O4+]| - содержит два остова и шесть электридов (восемь частиц). Важно подчеркнуть, что если координационное окружение остовов электридами соответствует координационным числам (КЧ) атомов соединения, то для всех электридов имеет место линейная координация остовами (КЧ = 2).
В результате уточнения и дополнения классической формулы ионной кристаллической решетки (Урусов, 1975, с. 23; Харрисон, 1983, с. 22), нами была выведена следующая формула энергии сцепления остовов и электридов для общего случая кристаллического соединения[3]:
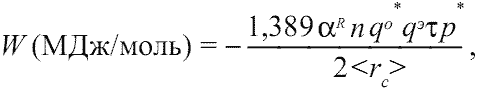 (1.9)
(1.9)
где: 1,389 - константа для выражения энергии сцепления остовов и электридов в МДж/моль[4]; αR - приведенная (связевая) константа Маделунга; n - суммарное число остовов и электридов, приходящихся на формульную единицу соединения; qo* - суммарный эффективный заряд ядра, действующий на валентные (сверх остовные) электроны атома; qэ - заряд электрида; τ - коэффициент отталкивания, равный 0,1¸0,7; p* - коэффициент, характеризующий ковалентность связей, равный (1-fi2)1/2, где fi - ионность соединения[5]; 2<rc>, (Å) - удвоенное среднее межатомное расстояние остов-электрид, равное удвоенным усредненным величинам ковалентных радиусов образующих кристалл атомов. Напомним, что в остовно-электронной модели ковалентный радиус атома равен сумме соответствующих ионных радиусов остова и электрида. Таким образом, для соединений МxXy, согласно схеме на рис. 1.3, можно записать: 2<rcM,X> = 2(rcM + rcX)/2 =
= rcM + rcX = d(M-X), где d -межатомное расстояние металл - неметалл в структуре. Следовательно, в знаменателе формулы (1.9) вместо члена 2<rc> можно поместить параметр d, Å среднего межатомного расстояния металл - неметалл соответствующего соединения:
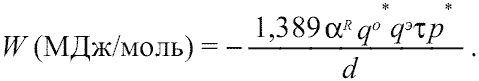 (1.10)
(1.10)
Очевидно, что в случае сложных кристаллических соединений в расчетах по формуле (1.10) необходимо использовать усредненные параметры qo*, qэ, fi, d.
Как указывалось, вместо номинального заряда остова (например, Na+, Zn2+, Al3+, C4+, S6+ и т. д.) в формуле (1.10) используется, согласно (Зуев, 1990; Зуев, Денисов, Мочалов и др., 2000), более адекватно характеризующий силовое поле остова параметр:
qo* = ΣZi*, (1.11)
где Zi* - эффективные заряды ядра, действующие на валентные электроны и рассчитываемые по формуле (Zhang, 1982):
Z* = n*(Iw/R)1/2, (1.12)
где n* - эффективное главное квантовое число, Iw (эВ) - потенциал ионизации атома соответствующей валентности (w), R - потенциал ионизации атома водорода, равный 13,595 эВ.
Например, расчеты по этим формулам дают для остовов:
[Na+] - qo* = 2,89(5,138/13,595)1/2 = 1,78;
[Zn2+] - qo* = 3,45(9,391/13,595)1/2 + 3,45(17,96/13,595)1/2 = 6,83;
[C4+] - qo* = 1,99(11,264/13,595)1/2 + 1,99(24,376/13,595)1/2 +
+1,99(47,86/13,595)1/2 + 1,99(64,48/13,595)1/2 = 12,54;
[S6+] - qo* = 2,89(10,357/13,595)1/2 + 2,89(23,4/13,595)1/2 +
+ 2,89(34,8/13,595)1/2 + 2,89(47,29/13,595)1/2 +
+ 2,89(72,5/13,595)1/2 + 2,89(88/13,595)1/2 = 30,35 и т. д.
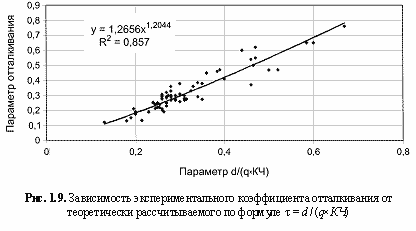
Необходимо объяснить сравнительно малую величину (0,1-0,7) коэффициента отталкивания, который в случае ионных кристаллических решеток гораздо выше (около 0,9). С одной стороны, это связано со значительным ростом положительных зарядов qo* по сравнению с соответствующими номинальными. С другой стороны, остовно-электронные модели кристаллов являются по образному выражению Бента (Bent, 1970) постройками из «ионов внутри ионов», что наглядно показано на примере периклаза на рис. 1.8. Этими обстоятельствами и объясняется столь сильное взаимное отталкивание одноименно заряженных частиц в остовно-электридных моделях. Обычно параметр t находится из данных по сжимаемости кристаллов. Нами для его оценки была выведена (рис. 1.9) следующая эмпирическая формула:
τ = 1,2656[d/(qxКЧ)]1.2, (1.13)
где d - межатомное расстояние в структуре; q - заряд электрида; КЧ - координационное число атомов (для сложных кристаллов все эти параметры являются средними для рассматриваемой структуры величинами).
Следует специально остановиться на оценке константы Маделунга αR, строгий вывод которой для сложных остовно-электронных моделей структур весьма трудоемок. Поэтому нами принят упрощенный метод ее расчета по формуле Темплтона (Урусов, 1975):
αR = 1,89 - 1/n x Σni/КЧi. (1.14)
Здесь ni - число частиц (остовов, электридов) i-сорта с соответствующими координационными числами; n - общее число остовов и электридов, приходящихся на формульную единицу соединения.
Например, для имеющего в своем составе два остова и четыре электрида сфалерита ZnS расчет по формуле (1.14) приводит к
αR = 1,89 - (2/4 + 4/2)/6 = 1,473. Для периклаза MgO (с двумя остовами и шестью электридами) получается такая же величина αR = 1,89 - (2/6 + 6/2)/8 = 1,473. Если (см. рис. 1.6) неподеленную пару электронов включить в остов атома кислорода, то остовно-электронная модель кварца будет |[Si4+](3e-)4[O4+]2|, и в этом случае αR = 1,89 - (1/4 + 2/2 + 4/2)/7 = 1,426.
Остановимся также подробнее на физическом смысле параметра р* в формуле (1.10). С этой целью рассмотрим ряд изоэлектронных и изоструктурных (типа сфалерита) соединений GeGe, GaAs, ZnSe, CuBr (таблица 1.10). Если обратиться к остовно-электронным схемам отдельных межатомных связей: [Ge4+](2e-)[Ge4+], [Ga3+](2e-)[As5+], [Zn2+](2e-)[Se6+] и [Cu1+](2e-)[Br7+] этих соединений, то они оказываются практически идентичными по всем кристаллохимическим параметрам (средним зарядам остовов, зарядам электридов, межатомным расстояниям и др.), по величинам энергий гипотетических неполярных (λ = 0) ковалентных связей, а также по величинам формульных весов, мольных объемов и плотностей. Тем не менее, в ряду CuBr→ZnSe→GaAs→GeGe имеет место явно ощутимый, закономерный рост термодинамической и механической стабильности соединений (фиксируемый соответствующим увеличением их энергоплотности Ev и относительной твердости НМ).
Таблица 1.10
Дата добавления: 2017-09-01; просмотров: 1100;
Поиск по сайту
Узнать еще
- H – параметры транзистора
- H-параметры транзистора
- H-параметры транзистора, включенного по схеме с ОБ.
- H-параметры транзистора, включенного по схеме с ОЭ.
- I. Энергетический блок.
- I.1.2 ТЕРМОДИНАМИЧЕСКАЯ СИСТЕМА. ТЕРМОДИНАМИЧЕСКИЕ ПАРАМЕТРЫ. УРАВНЕНИЕ СОСТОЯНИЯ
- I.2.1 ПОЛНАЯ И ВНУТРЕННЯЯ ЭНЕРГИЯ СИСТЕМЫ. ТЕПЛОТА И РАБОТА
- I.2.2 ТЕПЛОЁМКОСТЬ ВЕЩЕСТВА

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
