В СПЕКТРАХ КОМБИНАЦИОННОГО РАССЕЯНИЯ СВЕТА
Интенсивность линий комбинационного рассеяния (КР) света и спектры возбуждения линий КР, т.е. зависимость интенсивности от частоты возбуждающего света, можно вычислить с большими или меньшими ограничениями с использованием классической, полуклассической или квантовой теории [177, 212, 213]. Для проведения численных расчетов по любой из них необходимо знать величины многих параметров молекулы, часть из которых не удается получить из независимого эксперимента, но можно вычислить с помощью методов квантовой химии. Классическая теория, которую обычно называют "теорией поляризуемости", предназначена для случая большой удаленности от резонанса (частота возбуждающего света лежит далеко от полос поглощения). Интенсивность линий КР Ia в этой теории определяется совокупностью производных компонент тензора электронной поляризуемости молекулы ασρ по ядерной нормальной координате Qa. В случае линейно-поляризованного возбуждающего света (лазерное излучение) Ia = 9(ā2)' + 7(γ2)', где ā - сумма диагональных компонент;
γ - анизотропия тензора электронной поляризуемости. Значения компонент тензора электронной поляризуемости, необходимые для расчета Ia, могут быть вычислены во втором порядке теории возмущений по следующей формуле:
ασρ = (l/h) ∑e[(M0eσ*M0eρ)/(ve – v) + (M0eσ*M0eρ)/(ve + v)],
где M0eσ - декартова компонента дипольного момента электронного перехода 0—е;
ve - его частота; v - частота возбуждающего света. Кроме того, компоненты тензора ασρ могут быть вычислены с помощью теории конечных возмущений (см. ниже). Для расчета производных обычно используется метод численного дифференцирования.
В полуклассической теории использовано выражение для расчета Ia, такое же, как в классической; тензор ασρ вычисляется во втором порядке теории возмущений, но численное дифференцирование его компонент заменено дифференцированием компонент вектора дипольного момента электронного перехода и его энергии, которые входят в выражение для ασρ.
Согласно квантовой теории, интенсивность линий КР, соответствующая колебательному переходу 0→f пропорциональна
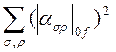
где α(σρ)0f - компоненты тензора электронной поляризуемости. При учете вкладов подуровней ev для каждой компоненты тензора можно написать равенство
α(σρ)0f = const ∑ev [<0|M0eσ|v> <v|M0eρ|f>/(vev — v) + <0|M0eρ|v> <v|M0eσ|f>/(vev + v)]. Здесь сумма берётся по всем электронно-колебательным переходам 00→ev, vev - их частоты. Квантовая теория может быть использована для расчета интенсивностей линий КР при любой частоте возбуждающего света как вдали, так и вблизи от резонанса с частотами электронно-колебательных переходов. В последнем случае к знаменателям vev — v необходимо добавить член iГev, учитывающий естественную ширину полосы поглощения перехода 00→ev.
Расчеты по теории поляризуемости интенсивности линий КР проведены для многих простых молекул (водорода, хлора, метана, этилена, этана, ацетилена, диметилового эфира и др.) [142, 146, 214]. Анализ результатов, полученных с использованием различных квантовохимических методов, показывает, что все они правильно, хотя и весьма грубо, передают распределение интенсивностей в спектрах КР, причем неэмпирические расчеты в базисах среднего размера типа 4-31ГФ не дают ощутимого повышения точности по сравнению с полуэмпирическими.
Более точные неэмпирические расчеты, в которых используются расширенные базисы с поляризационными орбиталями и учитывается электронная корреляция, приводят к очень хорошему согласию с экспериментом. Так, в работе [215] получены следующие значения производных поляризуемости для молекулы водорода (ат. ед.): a/||=178 и a/┴=0,83, опытные данные соответственно 1,69 и 0,85. Такое хорошее согласие с экспериментом не вызывает удивления, так как были получены [215] почти точные электронные волновые функции. К сожалению, для сложных молекул такие расчеты практически нельзя осуществить.
Основная трудность, которая возникает при использовании любой из перечисленных выше теорий для расчета интенсивности линий в нерезонансных спектрах КР света, заключается в необходимости вычисления параметров очень большого числа электронно-возбужденных состояний. Эту трудность удается обойти с помощью перехода к теории конечных возмущений [216], для расчетов по которой достаточно знания волновой функции только основного состояния. В этом приближении молекулу помещают во внешнее статическое электрическое поле и вычисляют, как при этом изменяется компоненты ее дипольного момента (Δμx, Δμy и Δμz)- Такие расчеты проводят с внешним полем, направленным по очереди вдоль каждой из трех декартовых осей координат, после чего с помощью численного дифференцирования рассчитывают компоненты тензора ασρ: ασρ = Δμσ/Eρ (σ, ρ = x, у, z).
Расчеты различными вариантами метода МО ЛКАО для основного состояния обычно дают более точные результаты, чем для электронно-возбужденных, поэтому можно ожидать, что использование теории конечных возмущений позволит вычислять с более высокой точностью значения интенсивностей линии КР при возбуждении вдали от резонанса. В работах [216 - 218] такие расчеты были проведены для метана, этилена, тетрахлорэтилена и других несложных молекул с использованием полуэмпирических и неэмпирических квантовохимических методов. Согласие с экспериментом получилось удовлетворительное.
Следует, однако, подчеркнуть, что с помощью теории конечных возмущений можно рассчитать лишь статические электронные поляризуемости, которые, вообще говоря, не имеют физического смысла, хотя могут немного отличаться от значений, найденных для небольших частот возбуждающего света v<<vev. Последнее оправдывает применение теории конечных возмущений для расчета спектров предельных углеводородов и некоторых других соединений, которые не содержат сопряженных связей и чьи электронные спектры поглощения лежат в дальней УФ-области.
Расчет интенсивностей линий КР этилена по полу классической теории проведен в работе [219]. В ней использован метод ППДП/С с учетом вкладов от всех однократно возбужденных конфигураций. Расчет с достаточно высокой точностью воспроизвел данные эксперимента при возбуждении линий КР в видимой области. При этом оказалось, что для линии полносимметричного валентного колебания связи С=С основной вклад в интенсивность дает длинноволновый электронный переход π→π*, а для остальных линий наиболее существенными являются вклады от более коротковолновых электронных переходов, которые локализованы преимущественно на связях С—Н. Были проведены расчеты интенсивностей линий КР и спектров возбуждения по квантовой теории для этилена и коротких полиенов и сделан ряд предсказаний [220 - 224].
В частности, показано, что у этилена при возбуждении в длинноволновой полосе поглощения, соответствующей π→π*-электронному переходу, наиболее интенсивными должны быть линии первого и третьего обертонов крутильного колебания [220]. Экспериментальных данных в то время не было, а в резонансных спектрах КР других молекул крутильные колебания и их обертоны не наблюдались. Однако в дальнейшем этот нетривиальный результат получил экспериментальное подтверждение [225].
Доступность для расчета электронных волновых функций и энергий возбужденных состояний в различных вариантах метода МО ЛКАО делает возможным привлечение этих методов при изучении практически любых молекулярно-оптических явлений. С их помощью для несложных соединений, комплексов, ионов, радикалов, молекулярных кристаллов можно предсказать общин вид спектров поглощения и излучения и дать отнесение наблюдаемым электронным переходам, составить суждение о зависимости спектра от конформации, начального электронного состояния и дополнительных возмущений. Вместе с тем можно уточнить и дополнить сведения о силовом поле, определить знаки производных поляризуемости и дипольного момента, оценить частоты ненаблюдаемых переходов, охарактеризовать кривые потенциальной энергии для возбужденных состояний и т.д. Точность, с которой можно рассчитать спектральные параметры органических соединений методами квантовой химии, в настоящее время значительно ниже точности эксперимента, однако достаточна для решения многих спектроскопических и спектрохимических задач. При этом совокупность данных расчета и эксперимента приводит к более полному и достоверному описанию спектра. Привлечение квантовохимических методов позволяет повысить информативность спектроскопических исследований, раскрыть основы эмпирически установленных закономерностей, связывающих спектры с различными физическими и химическими свойствами молекул, выяснить природу взаимного влияния функциональных групп в сложных соединениях, опираясь на представления о взаимодействии электронов и ядер.
ЗАКЛЮЧЕНИЕ
Прочитав настоящую книгу, читатель имел возможность убедиться, что с помощью квантовохимических расчетов можно получить много интересной информации о строении, физико-химических свойствах и реакционной способности органических соединений. Методы квантовой химии позволяют рассчитывать параметры любых молекул, интермедиатов и даже переходных состояний. Поэтому в настоящее время их широко применяют в химических исследованиях. Ежегодно публикуется более тысячи работ в этой области. Однако желающие использовать квантовохимические методы всегда должны помнить о приближенном характере как полуэмпирических, так и неэмпирических расчетов. Для неспециалиста название "неэмпирический" в настоящее время приобрело значение слова "точный". На самом же деле термин "неэмпирический" характеризует лишь способ вычислений матричных элементов, но не точность метода. Приближенный характер квантовохимических расчетов приводит к тому, что для вычисления различных параметров молекул приходится пользоваться разными методами, и правильный выбор их является важной частью работы. Лишь при рациональном и обоснованном выборе метода квантовохимические расчеты позволяют получить интересную информацию о реакционной способности органических соединений, дать объяснение экспериментально наблюдаемым закономерностям и даже сделать некоторые предсказания. При этом следует помнить, что получить надежный численный результат для каждой конкретной реакции квантовая химия в настоящее время не позволяет. С ее помощью обычно удается сделать лишь весьма общие заключения о механизме реакции и о влиянии на него различных факторов, а в отдельных случаях даже сформулировать новые концепции. В свою очередь, на основе этих представлений удается на качественном уровне предсказать некоторые параметры конкретных реакций. В настоящее время квантовохимические расчеты обычно проводят на больших ЭВМ. Это затрудняет их использование непосредственно в химических лабораториях. Однако очень быстро совершенствуются персональные компьютеры, и уже сейчас на некоторых моделях можно проводить квантовохимические расчеты, по крайней мере полуэмпирическими методами. В ближайшие годы такие компьютеры будут в арсенале большинства химиков-экспериментаторов и провести квантовохимические расчеты станет намного легче, поэтому их применение в органической химии будет расширяться, и мы надеемся, что эта книга поможет нашим читателям преуспеть в данной области.
ЛИТЕРАТУРА
1. Мак-Вини Р., Сатклиф Б. Квантовая механика молекул. М.: Мир, 1972. 380 с.
2. Дьюар М. Теория молекулярных орбиталей в органической химии. М.: Мир, 1972.
590 с.
Дата добавления: 2020-12-11; просмотров: 707;
Поиск по сайту
Узнать еще
- Анализ лучевого распространения света в волоконных световодах
- Атомный фактор рассеяния
- В) источники электрического света
- Взаимодействие света с границей раздела сред. Формулы Френеля
- Взаимодействие света со стеклом
- ВИДЫ ОСВЕЩЕНИЯ И ХАРАКТЕРИСТИКИ ИСТОЧНИКОВ СВЕТА
- Волновая оптика. Интерференция света

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
