ИССЛЕДОВАНИЕ ВОЗБУЖДЕННЫХ СОСТОЯНИЙ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Квантовохимические расчеты электронной структуры и геометрии электронно-возбужденных молекул широко используются для интерпретации экспериментальных данных и отнесения полос поглощения, установления связи спектров поглощения со структурой молекулы и ее конформацией, а также для изучения реакционной способности молекул в фотохимических реакциях.
Рассмотрим несколько примеров. В спектрах поглощения некоторых органических соединений характерные интенсивные длинноволновые полосы ранее предположительно относили к переходам с переносом заряда, но доказать это и оценить вклад конфигурации с переносом заряда в электронные переходы удалось только на основе квантовохимических расчетов. В частности, для N-алкил-N-нитрозоанилина и его алкилпроизводных установлено [192], что первая, длинноволновая, полоса поглощения относится к электронному переходу, локализованному на бензольном кольце (1Lb); возбуждение во второй полосе поглощения - к электронному переходу, локализованному на группе NNO; поглощение в третьей полосе — к электронному переходу с переносом заряда с группы NNO на бензольное кольцо (вклад этой конфигурации составляет около 50%); поглощение света в четвертой полосе - к электронному переходу, локализованному на бензольном кольце (1La). Из расчета также следует, что полоса с переносом заряда должна наблюдаться на опыте только у aнти-изомера, а у син-струк-туры она должна быть близка к более интенсивной полосе 1La и маскироваться ею.
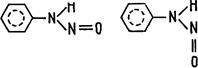
Син анти
Опыты показали, что некоторые несопряженные соединения с несколькими двойными связями, содержащие фрагмент типа
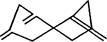
имеют интенсивную полосу поглощения около 230 нм, в то время как у моноолефинов первая интенсивная полоса поглощения расположена в области короче 200 нм. Неэмпирические расчеты привели авторов работы [193] к заключению, что эта длинноволновая полоса обусловлена электронным переходом между граничными π- и π*-орби-талями, энергетическая щель между которыми уменьшена за счет взаимодействия между двойными связями, разделенными метиленовыми мостиками.
При исследовании электронных спектров поглощения аллильных соединений ртути, олова и некоторых других тяжелых элементов СН2=СНСН2Х (X=HgH, SnH3, ...) были обнаружены ярко выраженные признаки неаддитивности [194 - 196], в частности длинноволновая полоса поглощения, которой нет у молекул СН2=СНСН3 и НХ, имитирующих в некотором смысле фрагменты аллильного соединения. Позднее значительные отклонения от аддитивности были обнаружены в фотоэлектронных спектрах [197 - 199]. Оказалось, что первые потенциалы ионизации аллильных соединений ртути и олова почти на 1 эВ меньше первых потенциалов ионизации фрагментов. В винительных соединениях типа СН2=СНХ и циклических структурах типа
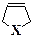
подобные эффекты не наблюдаются [196].
Для интерпретации этих экспериментальных результатов в работах [200, 201] были использованы результаты квантовохимических расчетов методом МПДП. Оказалось, что у всех исследованных аллильных соединений верхняя занятая МО антисимметрична по отношению к плоскости атомов С=С—С и локализована преимущественно на связи С=С, т.е. является π-орбиталью. Немного ниже нее, на шкале энергий, лежит σ-орбиталь, симметричная по отношению к плоскости атомов С=С—С и локализованная в значительной степени на группе с тяжелым атомом. Нижней вакантной МО является σ*-орбиталь, локализованная в основном на группе с тяжелым атомом, немного выше нее на шкале энергий лежит разрыхляющая π*-орбиталь связи С=С. Такой порядок уровней имеет место у плоских конформеров аллильных соединений.
При переходе к неплоскому гош-конформеру, который является наиболее стабильным, между π- и σ-орбиталями появляется взаимодействие, они смешиваются и энергетическая щель между ними увеличивается. Аналогично смешиваются и изменяют свое положение на шкале энергий вакантные π*- и σ*-орбитали. В результате этого энергии π- и π*-орбиталей повышаются, а энергии σ- и σ*-орбиталей понижаются (рис. 3.2). Таким образом, щель между граничными МО уменьшается, и эти приводит к уменьшению энергии длинноволнового перехода. Изменение положения верхней занятой МО на шкале энергий ведет к уменьшению первого потенциала ионизации.
Рис. 3.2. Положение на шкале энергий MO CH2=CHCH2HgH
Дата добавления: 2020-12-11; просмотров: 586;
Поиск по сайту
Узнать еще
- I тип реакций. Реакции, характерные для органических кислот.
- II. Реакции диазосоединений без выделения азота
- II. Темы рефератов, ориентированные на исследование и анализ методологических идей и концепций крупнейших представителей современной философии и естествознания.
- II. Тривиальные стандартные названия органических кислот
- III. Исследование личностных особенностей подростков
- Im7im9im11Исследование памяти
- VI. АГРАФИЯ. ИССЛЕДОВАНИЕ ПИСЬМА
- VII. АЛЕКСИЯ. ИССЛЕДОВАНИЕ ЧТЕНИЯ

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
