РАСЧЕТ ТЕПЛОВЫХ ЭФФЕКТОВ ОРГАНИЧЕСКИХ РЕАКЦИЙ
Величина теплового эффекта позволяет оценить термодинамическую возможность протекания химической реакции или отдельной элементарной стадии. В общем случае теплота реакции не коррелирует с ее скоростью, но в ряде однотипных органических реакций, в которых участвуют родственные соединения, такая корреляция иногда наблюдается. Поэтому данные о тепловых эффектах широко применяются для изучения реакционной способности органических соединений. Для большинства реакций экспериментальные данные о тепловых эффектах отсутствуют, поэтому квантово-химические расчеты этих величин получили широкое распространение. Следует, однако, отметить, что расчет тепловых эффектов для квантовой химии является весьма сложной задачей, так как эту величину необходимо знать с точностью до около 4 кДж/моль. Некоторые авторы называют ошибку в 4 кДж/моль "химической точностью" [39]. Как видно из предыдущего раздела, средняя ошибка квантовохимического расчета теплот образования органических соединений составляет около 25 кДж/моль. При расчете тепловых эффектов реакций ошибки еще больше. Этой точности недостаточно для решения прикладных задач, однако для однотипных реакций, в которых участвуют родственные соединения, относительную величину теплового эффекта квантовохимические расчеты передают значительно точнее. Для таких реакций нередко удается достичь "химической точности" и получить данные, которые помогут химику-экспериментатору при изучении реакционной способности органических соединений и механизмов сложных реакций.
Таблица 1.6 Теплоты реакций изомеризации (кДж/моль)
| Реакция | МПДП | Эксперимент |
| СН3С≡H → СН2=С=CH2 | ||
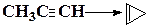
| ||
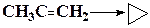
| ||

| ||
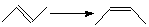
| ||
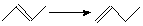
| ||
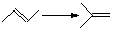
| -5 | |

| ||
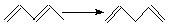
| ||
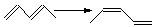
| ||
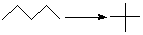
| -23 | |
| С2Н5NH2 → (СН3)2NH | ||
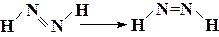
| ||
| СН3СN → СН3NС | ||
| С2Н5OH → СН3OСН3 | ||
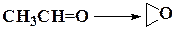
| ||
| СН3СOOН → НСOOСН3 | ||
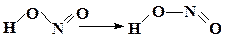
|
Для вычисления тепловых эффектов органических реакций применяют как полуэмпирические, так и неэмпирические квантово-химические методы. Из полуэмпирических наиболее широко используются схемы МЧПДП/3 и МПДП, причем метод МПДП дает более точные результаты. В табл. 1.6 приведены результаты расчетов этим методом тепловых эффектов некоторых реакций изомеризации. Из этих данных видно, что для некоторых реакций согласие теории с экспериментом хорошее, но в отдельных случаях, например для реакции СН3СN → СН3NС ошибка получается очень большой (около 100 кДж/моль). Поэтому при проведении прикладных расчетов необходимо первоначально на простых моделях убедиться, что для данной реакции или для данного класса реакций метод МПДП дает правильные результаты. Если ошибки получаются большими, но носят систематический характер, их можно устранить, введя соответствующие инкременты.
Результаты неэмпирических расчетов тепловых эффектов органических реакций очень сильно зависят от выбора метода [40-51]. В валентно-расщепленных базисах для реакций с участием насыщенных молекул ошибки составляют около 40 кДж/моль, но, если в молекулах есть кратные связи или молекулы являются напряженными, ошибки обычно увеличиваются приблизительно в 2 раза и достигают 80 кДж/моль [26, 40, 41].
Более точные результаты получаются для так называемых изодесмических реакций, т.е. для реакций, в которых число валентных связей каждого типа сохраняется, а изменяется лишь их взаимное расположение. Этому условию удовлетворяет, в частности, реакция ацетонитрила с метаном СН3СN + СН4 → СН3—СН3 + HCN.
При вычислении тепловых эффектов этих реакций с использованием базисов 3-21 ГФ и 4-31ГФ ошибки не превышают 20 кДж/моль [42—50]. Для неизодесмических реакций точность расчета существенно ниже. Особенно большие ошибки получаются при расчете тепловых эффектов реакций, в которых участвуют молекулы с малыми циклами, так как неэмпирический расчет в валентно-расщепленных базисах существенно недооценивает стабильность напряженных молекул по сравнению с соответствующими ациклическими соединениями [51]. Включение в базис поляризационных орбиталей уменьшает ошибку до 5-10 кДж/моль [42, 43], т.е. расчеты в базисах с поляризационными орбиталями позволяют вычислить тепловые эффекты реакции с почти "химической точностью". Исключение составляют реакции, в которых происходит гомолитический разрыв валентных связей. В этом случае ошибки при вычислении тепловых эффектов даже при использовании базисов с поляризационными орбиталями превышают 100 кДж/моль, что обусловлено необходимостью учета электронной корреляции [43—46].
В качестве примера использования данных квантовохимических расчетов тепловых эффектов органических реакций для изучения их механизмов рассмотрим результаты работы [52]. В ней был изучен механизм реакции 1,2,4-о-ацетил-α-D-ксилопиранозы со спиртами в условиях кислотного катализа.
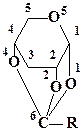
На основе анализа строения конечных продуктов этой реакции экспериментаторы предполагали, что на первой стадии происходит протонирование атома кислорода О1 и образуется ацилоксониевый ион следующего строения:
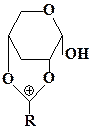
Этот ацилоксониевый ион далее вступает в реакцию с молекулами спирта. Однако дать ответ на вопрос, почему протонируется атом кислорода О1 и не протонируются атомы кислорода О2, О4 и О5, не удалось. Остался не выясненным также механизм превращения протонированной по О1 формы исходной молекулы в ацилоксониевый ион. Для решений этой задачи в работе [52] были привлечены методы квантовой химии. С их помощью были рассмотрены четыре возможных направления протонирования исходной молекулы: по атомам кислорода О1, О2, О4 и О5. Было показано, что при присоединении протона к атомам кислорода О1, О2 и О4 разрыв валентных связей соответственно С6—О1, С6—О2 и С6—О4 термодинамически выгоден и происходит без активационного барьера, при этом образуются соответствующие ацилоксониевые ионы. Протонирование идет преимущественно по атому кислорода О1, так как такое направление реакции является термодинамически наиболее выгодным. Таким образом, направление этой реакции можно объяснить термодинамическим фактором. На второй стадии реакции ацилоксониевый ион вступает в реакцию со спиртом. По данным квантово-химических расчетов [52] ее направление можно объяснить распределением электронной плотности в ацилоксониевом ионе.
В качестве второго примера использования данных квантово-химических расчетов тепловых эффектов реакций для изучения реакционной способности рассмотрим результаты работы [53]. В ней было изучено влияние заместителей на сродство к протону бензильных соединений. Согласно многочисленным экспериментальным данным, заместители, включающие систему связей —С—Х (X — атом тяжелого металла), повышают относительную стабильность карбкатионов с π-связями. В частности, заместители Y= (СН3)3РЬСН2 и РhСН2НgСН2 приводят к существенному смещению равновесия в пользу карбкатиона в следующей реакции:
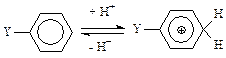
Для этих заместителей константы σ+p, характеризующие равновесие в данной реакции, достигают величины -1,1 (заметим для сравнения, что у такого заместителя, как СН3O—, с достаточно сильными π-электронодонорными свойствами, данная константа равна -0,75). Однако вопрос о механизме стабилизации карбкатионов оставался открытым. В связи с этим в работе [53] было проведено изучение электронного строения бензильных соединений кремния, германия, олова и свинца, а также их протонированных форм:
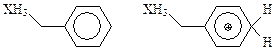
(X = Si, Ge, Sn, Рb).
Первоначально было показано, что квантово-химический расчет позволяет воспроизвести данные, известные из эксперимента, т.е. что по данным расчета сродство к протону возрастает при введении в молекулу заместителя с тяжелым атомом (табл. 1.7). После этого была выяснена причина увеличения сродства к протону. Оказалось, что при введении в молекулу заместителя с тяжелым атомом σ-орбитали сближаются с π-орбиталями по своему положению на шкале энергий. В результате у неплоских конформеров усиливается смешивание σ- и π-орбиталей. Это приводит к тому, что положительный заряд у протонированных форм соединений с тяжелыми атомами сильнее делокализован, а увеличение делокализации заряда приводит к стабилизации соединения. Таким образом, экспериментально наблюдаемый эффект является следствием смешивания σ- и π-орбиталей, или σ,π-сопряжения.
Таблица 1.7Сродство к протону (кДж/моль) бензильных соединений C6H5CH2XH3
| Х | Неплоский конформер | Плоский конформер |
| Si | 8,4 | 1,7 |
| Ge | 14,3 | 2,0 |
| Sn | 16,0 | 2,0 |
| Рb | 23,1 | 2,0 |
Примечание. За нуль принято сродство к протону этилбензола.
Таблица 1.8 Барьеры внутреннего вращения (кДж/молъ) в бензильных соединениях C6Н5CH2XH3 и их протонированных формах C6Н6CH2XH3 вокруг связи С—С между бензольным кольцом и метиленовой группой [53].
| Х | Бензильное соединение | Протонированная форма |
| С | ||
| Si | ||
| Ge | ||
| Sn | ||
| Рb |
σ,π-Сопряжение усиливается при переходе к бензильным соединениям с более тяжелыми атомами, причем в протонированных формах оно проявляется сильнее, чем в электрически нейтральных. Oб этом свидетельствуют данные табл. 1.8, в которой видно, что барьер внутреннего вращения при переходе от этилбензола к бензильным соединениям олова и свинца возрастает в 1,3—1,7 раза. Скорее всего, этот рост связан с увеличением размера атома Х и частично с усилением эффекта σ,π-сопряжения. В аналогичных карбкатионаx барьер внутреннего вращения возрастает в 2—4 раза. Столь значительное увеличение барьера не может быть связано с изменением стерических эффектов, так как они должны быть примерно одинаковыми в карбкатионах и электрически нейтральных молекулах; скорее всего, оно связано с усилением эффекта сопряжения за счет сближения σ- и π-орбиталей на шкале энергии при переходе к бен-зильным соединениям с тяжелыми атомами.
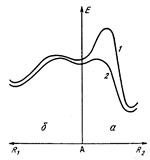
Рис. 1.3. Влияние заместителя на направление реакции образования 4,5-дигидрофуранов (а) и формилциклопропанов (б). А — исходный интермедиат; R1 и R2 — координаты реакций; 1 — R2=H или CH3; 2 — R2 = СНО.
Выше был приведен пример, когда направление реакции (протонирования 1,2,4-о-ацетил-α-D-ксилопиранозы) определяется величиной теплового эффекта. Однако на опыте далеко не всегда протекает термодинамически наиболее выгодная реакция. Как было указано, скорость реакции определяется свободной энергией активации и в общем случае не коррелирует с ее теплотой. Решая прикладные задачи, приходится достаточно часто сталкиваться с такими реакциями. В качестве примера рассмотрим результаты, полученные в работе [54]. Из эксперимента было известно, что карбанионы, генерированные в условиях межфазного катализа из броммалонового эфира, присоединяются к незамещенным или монозамещенным в β-положении α,β-непредельным альдегидам R1CH=C(R2)CHO, образуя или формилциклопропаны (R2=H,CH3), или формил-4,5-дигидро-фураны (R2=СНО) (см. схему реакций на стр. 35).
Однако выяснить причины такого влияния заместителей на направление реакции экспериментаторы не смогли. Поэтому для изучения ее механизма были привлечены методы квантовой химии. Расчет тепловых эффектов показал, что реакция, которая приводит к образованию конечных продуктов с пятичленным циклом, является термодинамически более выгодной и этот результат не зависит от заместителя. Действительно, формилциклопропаны содержат напряженный трехчленный цикл и поэтому являются термодинамически менее устойчивыми по сравнению с дигидрофуранами.
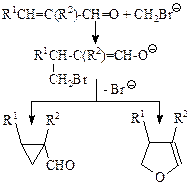
При детальном изучении механизма этой реакции было показано, что скорости двух конкурирующих процессов, которые приводят к образованию циклопропанов и дигидрофуранов, не коррелируют с их тепловыми эффектами, т.е. направление реакции определяется не ее термодинамикой, а кинетикой. Если R2=H или СН3, то на пути образования термодинамически более устойчивых дигидрофураиов лежит достаточно высокий активационный барьер. При комнатной температуре направление реакции определяется кинетическим фактором. В результате образуются термодинамически менее выгодные формилциклопропаны. При R2=CHO активационный барьер на пути образования 4,5-дигидрофуранов резко снижается и направление реакции определяется термодинамическим фактором, поэтому в результате реакции образуются более устойчивые формил-4,5-дигидрофураны (рис. 1.3).
Дата добавления: 2020-12-11; просмотров: 585;
Поиск по сайту
Узнать еще
- E) Расчет структурных составляющих очага деформации с одним нейтральным сечением
- I тип реакций. Реакции, характерные для органических кислот.
- I. Погрешности механической обработки. Точность обработки. Методы их расчёта
- II. Тривиальные стандартные названия органических кислот
- IV тип реакций. Реакции электрофильного замещения в ядре идут в соответствии с правилами замещения
- IV. РАСЧЕТ РЕКТИФИКАЦИОННЫХ КОЛОНН
- V. ПРИМЕР РАСЧЕТА ФИЛЬТРА ВЫСОКИХ ЧАСТОТ ЧЕБЫШЕВА
- А) Расчет электрической цепи методом свертывания

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
