РАСЧЕТ ПОТЕНЦИАЛОВ ИОНИЗАЦИИ
Потенциалы ионизации (ПИ) органических молекул обычно вычисляют по теореме Купманса, которая связывает ПИ k-го электрона (Ik) с энергией εk хартри-фоковской орбитали исходной молекулы, с замкнутой оболочкой: Ik= -εk. Для большинства соединений расчеты в этом приближении дают удовлетворительное согласие с экспериментальными вертикальными ПИ и поэтому широко используются для интерпретации данных фотоэлектронной спектроскопии. Кроме того, расчеты ПИ по теореме Купманса используются для изучения реакционной способности некоторых органических соединений (см. следующий раздел).
Наибольшее количество опубликованных расчетов ПИ выполнено методами МЧПДП/3 и МПДП. В табл. 1.3. приведены результаты расчетов ПИ методом МПДП. Из этих данных видно, что согласие с экспериментом получается хорошее. Для соединений с тяжелыми атомами точность расчета ПИ немного снижается. Для метода МПДП характерно заметное завышение ПИ. Так, для соединений, содержащих атомы брома, ПИ получаются завышенными в среднем почти на 1 эВ, а для соединений олова и иода — на 1,3 эВ. Однако эти ошибки носят систематический характер, и их удается уменьшить с помощью подбора соответствующих инкрементов [28, 29].
Таблица 1.3 Потенциалы ионизации, эВ
| Соединение | МПДП | Эксперимент | Отнесение* | Соединение | МПДП | Эксперимент | Отнесение* | |
| Метан | 13,9 30,6 | 14,0 22,9 | 1t2 1a1 | Аммиак | 32,9 | 27,0 | 1a1 | |
| Цианистый водород | 13,4 14,3 21,6 | 13,6 14,0 20,0 | 1π 3σ 2σ | |||||
| Этан | 12,7 13,3 15,1 24,8 | 12,1 15,0 20,4 | 1eg 2a1g 1eu 1a2u | |||||
| Азот | 14,9 16,2 21,1 | 15,6 17,0 18,8 | 2σg 1πu 1σu | |||||
| Этилен | 10,2 12,6 14,6 15,8 | 10,5 12,8 14,7 15,9 | 1b1u 1b1g 2ag 1b2u | |||||
| Вода | 12,2 14,5 19,1 40,0 | 12,6 14,7 18,5 32,2 | 1b1 2a1 1b2 1a1 | |||||
| Ацетилен | 11,0 15,9 21,0 | 11,4 16,4 18,7 | 1πu 2σg 1σu | |||||
| Диоксид углерода | 12,8 17,7 17,8 21,2 | 13,8 17,6 18,1 19,4 | 1πg 1πu 2σu 2σg | |||||
| Бензол | 9,4 12,5 12,6 14,4 15,2 16,8 17,5 | 9,2 11,5 12,3 13,8 14,7 15,4 16,9 | 1e1g 2e2g 1a2g 2e1u 1b2u 1b1u 2a1u | |||||
| Формальдегид | 11,0 14,2 16,3 16,9 | 10,9 14,4 16,0 16,8 | 2b2 1b1 2a1 1b2 | |||||
| Аммиак | 11,2 16,7 | 10,9 16,0 | 2a1 1e |
* Отнесение, сделанное на основе данных метода МПДП, для всех перечисленных в таблице соединений совпадает с общепринятым.
Для небольших соединений расчеты ПИ проводят неэмпирическими методами. При этом получают значения ПИ не только для валентных электронов, но и для электронов внутренних оболочек. Эти результаты можно использовать для интерпретации данных как фотоэлектронной, так и рентгеноэлектронной спектроскопии. Основной недостаток расчетов ПИ с использованием теоремы Купманса заключается в том, что в этом приближении не учитываются реорганизация электронной структуры и изменение геометрии, корреляционной энергии, а для соединений с тяжелыми атомами и релятивистских поправок при отрыве электрона. ПИ с учетом перечисленных выше эффектов можно вычислить по формуле I = Е(М) - Е(М+ •), где Е(M) и Е(М+ •) — полные энергии молекулы и ее катион-радикала. Если использовать геометрию исходной молекулы при вычислении полной энергии катион-радикала, то получится так называемый вертикальный ПИ. Однако у большинства органических соединений учет электронной релаксации незначительно меняет ПИ, кроме того, этот вклад частично компенсируется изменением корреляционной энергии, поэтому расчеты в приближении Купманса широко используются в прикладных работах.
Кроме вертикального ПИ, можно рассчитать и адиабатический (учетом изменения геометрии молекулы при ионизации). Для этого вычисление Е(М+) следует проводить с оптимизацией геометрии. Для некоторых соединений вертикальные и адиабатические ПИ сильно отличаются. Так, для молекулы метана вертикальный ПИ больше адиабатического на 1,7 эВ, для силана — на 1,2 эВ. Данные по вертикальным и адиабатическим ПИ используются при анализе колебательной структуры линий в фотоэлектронных спектрах (см. гл. 3) и для изучения механизмов ионизации органических соединений.
В качестве примера рассмотрим результаты работы [30], в которой было изучено влияние механических деформаций на образование заряженных частиц в полимерах. Из эксперимента известно, что при очень сильном сжатии в сочетании со сдвигом происходит спонтанная ионизация полимера, т.е. ПИ сильно деформированного полимера снижается до нуля. Механизм этого эффекта был рассмотрен [30] на примере молекулы метана.
На рис. 1.1 приведены результаты расчета теплот образования (полных энергий) метана и его катион-радикала в зависимости от угла α, а также зависимость ПИ от этого угла. Из этих данных видно, что потенциальная кривая для молекулы метана является почти параболой. Энергия деформации быстро возрастает с отклонением величины α от равновесного значения 109,5°. Для катион-радикала метана потенциальная кривая имеет качественно другую форму, которая обусловлена эффектом Яна—Теллера. Угол α, равный 109,5°, соответствует локальному максимуму полной энергии, а углы 60 и 160° - минимумам. Особо отметим, что вычисленный профиль потенциальной энергии для катион-радикала имеет существенно более пологую форму по сравнению с аналогичной кривой для молекулы метана.
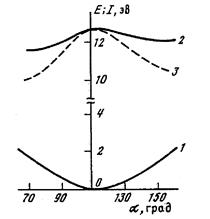
Рис. 1.1. Зависимость полных энергий метана (1) и его катион-радикала (2), а также зависимость ПИ метана (3) от угла Н—С—Н.
За нуль принята полная энергия равновесной конформации метана.
Приведенные результаты на первый взгляд противоречат нашей интуиции, однако их легко объяснить на основе анализа симметрии занятых электронами МО. Молекула метана в равновесном состоянии имеет симметрию Td, и ее верхняя занятая МО является трижды вырожденной. При удалении с нее электрона (без учета геометрической релаксации) образуется катион-радикал с трижды вырожденным основным состоянием. По теореме Яна—Теллера вырожденные состояния у нелинейных соединений неустойчивы и энергетически выгодными являются деформации геометрии, которые понижают их симметрию. Деформация валентных углов катион-радикала метана понижает его симметрию и, следовательно, энергетически выгодна, т.е. понижает его полную энергию. Из-за этого ПИ молекулы метана при деформациях, которые понижают ее симметрию, снижается.
Данный результат можно сформулировать в более общем виде: если верхняя занятая МО у молекулы вырождена или почти вырождена, то механические деформации, которые снижают симметрию молекулы и снимают вырождение, приводят к уменьшению ее ПИ. В случае вырожденных МО этот вывод обусловлен эффектом Яна—Теллера, а в случае почти вырожденных МО — псевдоэффектом Яна—Теллера. Уменьшение ПИ приблизительно равно энергии механической деформации в основном состоянии.
Аналогичный результат был получен и для пропана, причем было показано, что при механической деформации молекулы, когда изменяется несколько геометрических параметров, в уменьшение ПИ вносит вклад суммарная энергия деформации. В этом заключается качественное различие в механизмах влияния деформаций на ионизацию и на деструкцию (разрыв валентных связей) полимера. Например, для разрыва макромолекулы полиэтилена требуется сосредоточить на одной валентной связи С—С энергию 3,5 эВ, что при квазиоднородном растяжении достаточно большого участка цепи макромолекулы требует десятки электрон-вольт упругой энергии. Для ионизации полиэтилена необходимо затратить заметно больше энергии, чем на разрыв одной связи С—С (потенциал ионизации полиэтилена составляет около 7 эВ), но эта энергия может быть рассредоточена по нескольким степеням свободы, так как в снижение ПИ вносит вклад суммарная энергия деформации фрагмента полимерной цепи. Поэтому ионизация может происходить легче, чем деструкция.
Дата добавления: 2020-12-11; просмотров: 586;
Поиск по сайту
Узнать еще
- E) Расчет структурных составляющих очага деформации с одним нейтральным сечением
- I. Погрешности механической обработки. Точность обработки. Методы их расчёта
- IV. РАСЧЕТ РЕКТИФИКАЦИОННЫХ КОЛОНН
- V. ПРИМЕР РАСЧЕТА ФИЛЬТРА ВЫСОКИХ ЧАСТОТ ЧЕБЫШЕВА
- А) Расчет электрической цепи методом свертывания
- А. Расчет на устойчивость
- АВТОМАТИЗАЦИЯ МЕЖБАНКОВСКИХ РАСЧЕТОВ
- Автоматизация расчета и построения базовых конструкций одежды

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
