КЛАССИФИКАЦИЯ МОДЕЛЕЙ
Большинство органических реакций протекает в жидкой фазе. Очень часто растворитель изменяет не только выходы конечных продуктов, но и механизм реакции. В газовой фазе многие реакции идут по радикальному механизму, а в растворе - по ионному. Поэтому при квантовохимическом изучении реакционной способности органических соединений необходимо учитывать взаимодействие со средой.
В основе современных представлений о взаимодействии растворенных соединений с растворителем лежат ставшие классическими работы Борна, Дебая, Леннард-Джонса, Лондона, Кирквуда и Онзагера. Эти работы, их дальнейшее развитие и роль в химии подробно проанализированы в монографии [84]. Однако использовать непосредственно приведенные там результаты для учета сольватации в квантовохимических расчетах не удается, поэтому были разработаны более простые модели, которые позволяют рассчитать энергию сольватации с достаточно высокой степенью точности. Прежде чем перейти к описанию этих моделей, рассмотрим возможные типы сольватации.
Сольватацию принято разделять на два типа - специфическую и неспецифическую. Для первой характерно существование в растворах структурно-определенных образований между сольватом и сольвентом, время жизни которых заметно превышает период свободного колебания этой выделенной системы. Примерами подобных образований являются водородные связи, комплексы с переносом заряда и т.д. Для всех них характерны структурная определенность и относительно большая величина энергии взаимодействия, которая может иногда превышать 10% от энергии химической связи, а также существенное перераспределение заряда между растворенным соединением и растворителем.
Несколько особняком лежит случай сольватации малых одно-, двух- и некоторых трех- и четырехатомных ионов. Для этих соединений характерны все перечисленные особенности, но связь оказывается необычайно прочной. Так, энергия гидратации ОН - составляет 445 кДж/моль, Н3О+ 382 кДж/моль, NH3 344 кДж/моль. Такие структуры, по-видимому, было бы правильнее рассматривать как координированные.
В случае неспецифической сольватации энергия взаимодействия между растворенным соединением и молекулами растворителя невелика и не может создать сольватационные оболочки с жестко-фиксированной структурой. Основной вклад в энергию сольватации этого типа вносят ван-дер-ваальсово взаимодействие и электростатическое взаимодействие дипольных моментов растворенного соединения и молекул растворителя.
Методы квантовой химии, которые в настоящее время используются для описания сольватационных эффектов, можно разделить на две большие группы, дискретные и континуальные. Дискретные подходы обычно имеют дело с описанием какого-либо выделенного объема системы растворенное соединение-растворитель с включением до десяти молекул последнего. Эти молекулы располагают вокруг растворенного соединения в определенной системе (приближение "супермолекулы", различные варианты модели точечных диполей или зарядов). Подобное описание близко к химическому определению специфической сольватации. Они различаются лишь тем, что в расчет частично включается неспецифическое взаимодействие растворенного соединения и растворителя.
В некоторых работах рассматривались дискретные системы, содержащие 200-300 молекул растворителя (метод Монте-Карло с атом-атомными потенциалами). Такие большие кластеры пригодны для описания как специфического, так и неспецифического взаимодействия растворенного соединения с растворителем. Однако расчеты для них связаны с очень большими затратами машинного времени. Поэтому для учета неспецифической сольватации широкое распространение получили континуальные модели (сольватонная модель, модель самосогласованного поля реакции). В них не учитывается микроскопическая структура растворителя, поэтому отпадает необходимость определять строение сольватационной оболочки. Благодаря этому расчет становится существенно проще.
Полную энергию молекулы в растворителе можно представить как сумму двух отдельных вкладов: Еполн= ЕМ + ЕС ,
где ЕМ — энергия изолированной молекулы; ЕС — энергия сольватации. В предположении отсутствия специфического взаимодействия между растворенным соединением и растворителем, т.е. существенного перераспределения заряда между ними (этот случай будет рассмотрен отдельно), ЕС является суммой трех вкладов:
ЕС = Еэл + Едисп + Екав.
где Еэл - энергия электростатического взаимодействия между собственными и наведенными зарядами растворенного соединения и молекулами растворителя;
Едисп - дисперсионная составляющая энергии взаимодействия, учитывающая ван-дер-ваальсово взаимодействие; Екав - так называемая кавитационная энергия, т.е. энергия реорганизации растворителя, необходимая для образования полости, в которой помещается растворенное соединение. Расчеты с учетом всех трех вкладов в энергию сольватации показали, что для наиболее интересных полярных и заряженных систем в полярных растворителях, когда сольватация может существенно изменить результаты газофазных расчетов, Еэл превышает Едисп и Екав; кроме того, оказалось, что именно Еэл существенно изменяется в ходе реакции, а небольшие изменения Едисп и Екав в значительной мере компенсируют друг друга. Это привело к разработке ряда моделей, в которых сольватация учитывается в электростатическом приближении (различные варианты модели точечных диполей или зарядов, большинство континуальных моделей).
В настоящее время предложено много разных способов учета сольватации. Ниже будут рассмотрены лишь те из них, которые получили наиболее широкое распространение.
2.2. СОЛЬВАТОННАЯ МОДЕЛЬ
Эта модель была предложена Джано [85] и Клопманом [86]. В ее основе лежит предположение, что с каждым атомом растворенного соединения связан индуцированный в растворителе электрический заряд - сольватон. Заряд сольватона по абсолютной величине равен заряду на атоме, который его индуцировал, но имеет противоположный знак. Сольватоны не взаимодействуют между собой. Энергия взаимодействия между сольватоном С и атомом А вычисляется по следующей формуле:
EСА=qCqAе2(ε - 1)/(2rε)
где r — радиус атома А, если сольватон С индуцирован зарядом данного атома, или расстояние между атомами А и В, если сольватон индуцирован зарядом атома В;
qC и qА — заряды сольватона С и атома А в единицах заряда электрона;
е - заряд электрона; ε - диэлектрическая проницаемость среды.
Чаще для оценки ЕСА используют аналогичное выражение с кулоновскими интегралами, взятыми из квантовохимического расчета, т.е. е2/r полагается равным γАА, если сольватон С индуцирован зарядом на атоме А, или γАB, если сольватон С индуцирован зарядом атома В (γАA и γАB — одно- и двухцентровые кулоновские интегралы соответственно; см.разд. 1.2). Для энергии сольватации в этом приближении можно записать следующее выражение, где сумма берется по всем атомам растворенной молекулы:
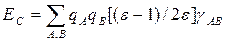
В качестве примера использования сольватонной модели для изучения механизмов органических реакций рассмотрим результаты работы [87], в которой изучена Z,E-изомеризация α,β-дизамещенных α,β-непредельных альдегидов.
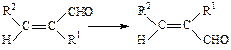
Расчеты были проведены методами ППДП/2 и МЧПДП/3. Эта реакция связана с внутренним вращением вокруг двойной связи С=С и в газовой фазе идет с очень высоким активационным барьером 200 - 250 кДж/моль. Преодолеть такой активационный барьер при нормальных условиях очень трудно, поэтому скорость газофазной реакции будет ничтожно мала.
Для реакции в растворе можно предложить следующие два механизма катализа, которые позволяют снизить активационный барьер.
1. Если растворитель обладает кислотными свойствами, то становится возможной протонизация исходного соединения, после чего по данным квантохимических расчетов активационный барьер реакции должен уменьшиться до 105 - 110 кДж/моль.
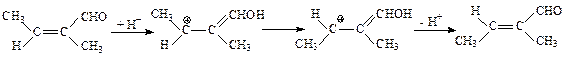
Учет влияния растворителя с использованием сольватонной модели (величина ε полагалась равной 5 и 10) приводит к дополнительному понижению высоты активационного барьера, и он становится равным 75 - 85 кДж/моль. Реакции с таким активационным барьером могут проходить при нормальных условиях. Таким образом, если растворитель обладает кислотными свойствами, то он может существенно ускорить реакцию Z,E-изомеризации α,β-дизамещенных α,β-непредельных альдегидов.
2. Если растворитель способен катализировать перенос атома Н от группы СН3 к СО, то становится возможным второй механизм увеличения скорости изомеризации:
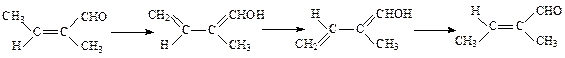
Высота активационного барьера изомеризации, проходящей по второму механизму, определяется высотой активационного барьера переноса протона от группы СН3 к карбонильному кислороду. Эта реакция, согласно результатам квантовохимических расчетов в газовой фазе, требует затраты около 65 кДж/моль. Учет сольватации повышает эту величину до 80 кДж/моль. Если растворитель обладает хорошей способностью к переносу протона (такой же, как вода), то высота активационного барьера будет близка к тепловому эффекту реакции, т.е. активационный барьер будет немногим выше 80 кДж/моль. С таким активационным барьером реакция может проходить при нормальных условиях.
Сольватонная модель проста и экономична. Однако она обладает рядом серьезных недостатков. Самый неприятный из них заключается в том, что сольватоны локализуются в непосредственной близости от атомов растворенной молекулы, которые их индуцировали, т.е. фактически проникают внутрь молекулы. В случае ионов сольватонная модель учитывает изменение энергии сольватации только за счет изменения степени делокализации заряда, хотя в действительности определяющим фактором в данном случае является их объем. Если изменение степени делокализации заряда коррелирует с изменением объема иона, то сольватонная модель будет давать правильные результаты. Из этого становятся ясными ограничения области ее применения. В частности, сольватонная модель не может учесть эффекты экранирования заряда неполярными группами или молекулами реагентов. По этой причине ее нельзя применять в комбинации с приближением "супермолекулы" при необходимости учесть эффект специфической сольватации.
В заключение настоящего раздела отметим еще одно важное обстоятельство. Выше было указано, что заряды на атомах, вычисленные разными методами, сильно различаются. Из-за этого сольватонная модель, будучи примененной в рамках разных методов, дает разные результаты. Она неплохо работает с методом ППДП/2. Мы пробовали использовать ее также в расчетах методами МЧПДП/2, МЧПДП/3 и МПДП, но результаты получились гораздо хуже.
2.3. МОДЕЛЬ САМОСОГЛАСОВАННОГО ПОЛЯ РЕАКЦИИ
В рамках макроскопической теории (шарик с фиксированным распределением зарядов погружен в среду с диэлектрической проницаемостью ε, заряды локализованы внутри шарика) энергия взаимодействия с растворителем задается электростатическим классическим потенциалом
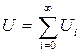 , где U0=(q2/2α)(1– 1/ε); U1=(μ2/2α3)[2(ε – 1)/(2ε + 1)]
, где U0=(q2/2α)(1– 1/ε); U1=(μ2/2α3)[2(ε – 1)/(2ε + 1)]
и т.д. Здесь q, μ и α - заряд, дипольный момент и радиус шарика соответственно. Первый член этого ряда называют борновским, второй - онзагеровским. Следующие члены ряда зависят от квадрупольного и более высоких мультипольных моментов шарика. Для электрически нейтральных шариков Uo=0 и первым ненулевым членом ряда будет U1. В работах Тапиа с сотр. [88 - 92] предложено добавить аналог члена U1 к гамильтониану изолированной молекулы и учесть таким способом взаимодействие с растворителем. В результате ими было получено следующее выражение для вычисления гамильтониана молекулы, находящейся в растворителе:
 Ĥ=Ĥ0 -
Ĥ=Ĥ0 - 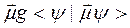
где g - тензор, который определяется восприимчивостью электростатического поля молекулы средой. В работах [88 - 92] он задавался в параметрической форме.
Этот метод учета взаимодействия с растворителем получил название модели самосогласованного поля реакции. Основные его недостатки очевидны. Они заключаются в необходимости учета зависимости тензора g от значения диэлектрической проницаемости растворителя и размеров молекулы. Это нельзя сделать без очень грубых дополнительных предложений. Обычно полагают, что диагональные элементы g равны 2(ε - 1)/[(2ε + 1)α3], а недиагональные - нулю. Однако перенесение макроскопической формулы на микроскопический уровень, вообще говоря, неверно, диэлектрическая проницаемость при этом теряет свой физический смысл, а выбор значения параметра α (радиуса молекулы) достаточно произволен. Поэтому модель самосогласованного поля реакции имеет ограниченную область применения. Практически ею можно пользоваться при рассмотрении реакций изомеризации и переноса протона. В остальных случаях ее использование нежелательно.
2.4. ПРИБЛИЖЕНИЕ СУПЕРМОЛЕКУЛЫ
С точки зрения построения модели наиболее простым способом учета сольватации является включение большого числа молекул среды в систему, для которой проводится квантовохимический расчет. Все ее электроны (сольватируемой молекулы и молекул среды) включаются в электронный гамильтониан. Этот способ является непосредственным обобщением квантовохимических методов, развитых для отдельных молекул на случай больших систем, состоящих из нескольких или даже большого числа отдельных молекул. Если нам удастся учесть таким способом взаимодействие растворенной молекулы с большим числом молекул cреды, рассчитать энергетически наиболее выгодную конформацию растворенной молекулы и конфигурацию молекул растворителя и получить для этой конфигурации электронную волновую функцию, то мы сможем объяснить или даже предсказать практически все интересующие нас свойства молекулы в растворе. При этом следует с особой осторожностью подходить к выбору квантовохимического метода, к которому в этом случае предъявляются повышенные требования, а именно он должен быть пригоден для изучения межмолекулярных взаимодействий.
Из предыдущей главы было видно, что многие квантовохимические методы, которые успешно используются для изучения реакционной способности органических соединений дают неправильные результаты при расчете параметров, характеризующих межмолекулярные взаимодействия. Например, метод МПДП дает неудовлетворительные результаты при расчете систем с водородными связями. Поэтому при использовании приближения "супермолекулы" для учета сольватации приходится выбирать достаточно совершенный квантовохимический метод. При этом возникают трудности, связанные с очень большим размером супермолекулы, которая состоит из растворенного соединения и молекул среды. Кроме того, в супермолекуле обычно неизвестно большое число геометрических параметров, которые определяют строение сольватационной оболочки. Поэтому выполнить реально такие расчеты удается только для систем с небольшим числом молекул среды.
Такой учет сольватации, при котором система из сольватируемой молекулы и некоторого ограниченного числа молекул растворителя рассчитывается квантовохимическим методом как одна молекула, получил название "приближения супермолекулы" [93, 94]. Расчеты в этом приближении широко распространены. К ним относятся работы, в которых вычисляются параметры для комплексов, состоящих из растворенной молекулы и одной молекулы растворителя, а также расчеты для комплексов с двумя, тремя и т.д. молекулами среды. В этих работах впервые удалось получить данные о строении и энергии взаимодействия для гидратных оболочек ионов ОН- и Н3О+ [65]. Несколько позже аналогичными методами исследованы гидратационные оболочки простейших ионов: Li+, Be2+, Na+, Mg2+, Al3+, К+, Ca2+, F-, Сl-, NH4+ [95 - 102], CH5+, CH5- [103], алкиламмониевых ионов [104].
Методика проведения перечисленных выше работ заложила основу подхода к изучению сольватации в приближении супермолекулы. Суть этих подходов заключается в следующем. Вначале рассчитывается энергия взаимодействия иона с одной молекулой воды и определяется наиболее энергетически выгодная конформация, потом в систему добавляется еще одна молекула воды и вновь рассчитываются энергия взаимодействия и структура комплекса и т.д. В результате такого расчета получается набор величин, которые являются энергиями гидратации иона А каждой последующей молекулой воды (En,n-1):
А•(n - 1)Н2O + Н2O → А•nН2O + En,n-1.
Для простейших ионов эти энергии были сопоставлены с экспериментальными данными, полученными методами масс-спектрометрии высоких давлений и циклотронного резонанса. Хорошее согласие с экспериментом подтвердило широкие возможности квантовохимических расчетов для изучения сольватации. Кроме того, были получены данные о числе молекул воды в первой гидратационной сфере и их конфигурация.
При переходе к более сложным молекулам расчеты в приближении супермолекулы существенно усложняются, так как центров сольватации уже много и заполнить даже всю первую гидратационную оболочку практически не удается. Поэтому в большинстве работ придерживаются следующей последовательности:
1) детально изучается взаимодействие сольватируемой молекулы с одной молекулой растворителя и получается предварительное представление о строении сольватационной оболочки;
2) изучается влияние второй, третьей и т.д. молекул растворителя на строение сольватационной оболочки и уточняются данные, полученные на первом этапе;
3) в изучаемую систему вводится максимально возможное количество молекул растворителя, для которого удается выполнить расчет, и для такой супермолекулы вычисляются все интересующие величины.
Детальное изучение взаимодействия сольватируемого соединения и одной молекулы растворителя включает их сближение (с различной взаимной ориентацией) и вращение вокруг собственных локальных осей. На этом этапе получают данные об основных наиболее выгодных положениях молекул растворителя, энергиях взаимодействия в таких положениях, расстояниях между растворенным соединением и молекулами растворителя и о подвижности последних. Обычно различают три вида подвижности: в положении равновесия, вблизи положения равновесия и вдали от положения равновесия.
Подвижность в положении равновесия — возможность молекулы растворителя вращаться вокруг собственных локальных осей. Для изучения этого вида подвижности рассчитывают зависимость энергии взаимодействия растворенного соединения и молекулы растворителя от углов поворота последней вокруг ее локальных осей. Расстояние между ними при этом не меняется. Подвижность вблизи положения равновесия — возможность молекулы растворителя смещаться на небольшие расстояния от положения равновесия. Для изучения этого вида подвижности рассчитывают форму ППЭ при небольших удалениях молекулы растворителя от положения равновесия без разрыва водородных связей, которые были в положении равновесия. Подвижность вдали от положения равновесия — возможность молекулы растворителя смещаться на большие расстояния от положения равновесия. Для изучения этого вида подвижности рассчитывают ППЭ при больших удалениях молекулы растворителя от положения равновесия, когда водородные связи между ней и растворенным соединением разорваны.
На втором этапе увеличивают число молекул растворителя в супермолекуле. При этом используют данные о положениях локальных минимумов, полученные на первом этапе, и лишь уточняют их положение. На третьем этапе в супермолекулу включают максимально возможное количество молекул растворителя (исходя из возможностей ЭВМ) и проводят расчет того или иного параметра.
В качестве примера рассмотрим результаты работы [105], в которой изучено строение гидратационной оболочки мочевины. Это соединение обладает рядом уникальных свойств. Так, в его присутствии резко снижается температура, при которой в водных растворах происходит денатурация белков и других биополимеров, повышается растворимость неэлектролитов и т.д. Принято говорить, что в присутствии мочевины разрушаются водородные связи между молекулами воды, другими словами, молекулы воды в водном растворе становятся более подвижными. Это приводит к перечисленным выше химическим и биохимическим эффектам. Однако механизм этого явления на микроскопическом уровне установлен не был. Для решения этого вопроса было изучено [105] строение гидратационной оболочки мочевины с помощью квантовохимических расчетов методами ППДП/2 и ОСТ-ЗГФ.
На первом этапе был проведен расчет строения комплекса, образованного одной молекулой мочевины с одной молекулой воды, и рассмотрена подвижность последней в этом комплексе. Оказалось, что наиболее устойчивый комплекс имеет следующую конфигурацию:
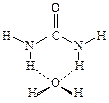
В этом комплексе молекула воды связана двумя водородными связями с двумя атомами Н связей N—Н, которые находятся в транс-положении по отношению к карбонильной группе. Расчет также показал, что в этом положении молекула воды обладает необычайно высокой подвижностью. Она может смещаться на расстояния до 0,15—0,20 нм от положения равновесия, и при этом энергия взаимодействия в комплексе почти не меняется. Таким образом, было обнаружено, что в исследованном комплексе молекулы воды и мочевины частично сохраняют свободу независимых перемещений в пространстве.
На втором этапе было увеличено количество молекул воды в комплексе, однако оказалось, что вторая молекула Н2О не может связаться с молекулой мочевины аналогично первой и образовать комплекс типа
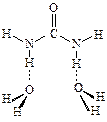
Таким образом, первая молекула воды, присоединяясь к мочевине, попадает в широкую потенциальную яму (именно этим объясняется ее высокая подвижность), но вторая молекула воды в эту потенциальную яму попасть не может, поэтому при увеличении количества молекул воды в комплексе высокая подвижность одной из них сохраняется. По-видимому, наличие этой достаточно широкой и глубокой потенциальной ямы обусловливает способность мочевины "разрушать структуру воды".
Введение мочевины в воду приводит к тому, что часть молекул воды, которые в чистом водном растворе (без мочевины) образовывали упорядоченную структуру (за счет образования водородных связей между собой), попадают в широкую потенциальную яму, созданную мочевиной. Такие молекулы не занимают определенного положения в пространстве и сохраняют способность перемещаться на большие расстояния (в пределах размеров потенциальной ямы). Поэтому как сами эти молекулы, так и молекулы воды из их непосредственного окружения не могут участвовать в образовании упорядоченной структуры воды. Подвижность молекул воды в данном случае увеличивается. Происходит как бы локальное повышение термодинамической температуры. Однако в действительности этот эффект обусловлен не повышением энергии молекулы воды, локализованной на ее вращательных и поступательных степенях свободы, а снижением потенциальных барьеров на пути ее перемещения.
Таким образом, предложен [105] механизм повышения термодинамической температуры водных растворов при добавлении в них мочевины. Квантовохимические расчеты помогли его найти, но сами по себе они не могут служить доказательством, что экспериментально наблюдаемые эффекты происходят именно по такому механизму. Для этого необходимо на основе предложенного механизма сделать предсказания, которые можно проверить экспериментально. Такие предсказания были сделаны [105]. Они касались влияния алкильных заместителей на способность мочевины повышать термодинамическую температуру водных растворов.
У метилмочевины наиболее устойчивой является следующая конформация:
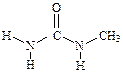
Наличие двух связей N—Н в транс-положении по отношению к карбонильной группе ведет к сохранению особенностей строения комплекса, описанных выше. Поэтому у метилмочевины сохраняется способность повышать термодинамическую температуру воды.
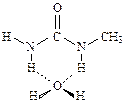
У диметилмочевины возможны два изомера
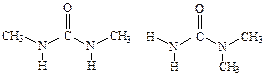
У первого из них две связи N—Н в транс-положении по отношению к карбонильной группе, а у второго - лишь одна. Следовательно, первый изомер будет повышать термодинамическую температуру воды и приводить к тем же эффектам, что и мочевина, а второй - нет. Это теоретическое предсказание получило экспериментальное подтверждение.
Приближение супермолекулы позволяет решать многие задачи, связанные с влиянием сольватации на реакционную способность органических соединений. Однако при его использовании необходимо самое серьезное внимание уделить выбору метода расчета. При этом следует руководствоваться двумя требованиями: 1) метод расчета должен быть достаточно точным и хорошо передавать как основные свойства растворенной молекулы, так и строение сольватационных оболочек; 2) расчет в приближении супермолекулы связан с вычислением электронной волновой функции для очень большой системы, и он должен быть практически реализуем исходя из возможностей имеющихся ЭВМ, их быстродействия и памяти. До недавнего времени расчеты с учетом сольватации в приближении супермолекулы проводились методами ППДП/2 и ОСТ-ЗГФ. Именно эти методы были использованы в приведенном выше примере изучения особенностей строения гидратационной оболочки мочевины. В настоящее время предпочтительнее использовать метод МПДП/Н и неэмпирические расчеты в базисе 3-21ГФ.
2.5. МЕТОД АТОМ-АТОМНЫХ ПОТЕНЦИАЛОВ
В приближении супермолекулы мы рассматривали систему из сольватированного соединения и некоторого числа молекул растворителя как одну большую молекулу. Такой подход является непосредственным обобщением методов квантовой химии, разработанных для расчета свойств отдельных (изолированных) соединений на межмолекулярные взаимодействия. При этом в качестве исходных частиц приходится оперировать электронами и атомными ядрами. При изучении системы, состоящей из одной молекулы, такой подход является единственно возможным, так как только на этом уровне можно анализировать большинство химических свойств молекулы. При рассмотрении межмолекулярных взаимодействий появляется возможность оперировать не с электронами и атомными ядрами, а с отдельными молекулами. Для этого необходимо иметь потенциалы, описывающие межмолекулярные взаимодействия. В случае системы, состоящей из электронов и ядер, необходимо решать уравнение Шредингера, так как электроны следует рассматривать как квантовые частицы. При рассмотрении межмолекулярных взаимодействий молекулы можно рассматривать как классические объекты. Благодаря этому появляется возможность использовать для их описания эмпирические потенциальные функции. Это существенно упрощает задачу.
Различные эмпирические потенциалы, которые предлагались различными авторами для описания межмолекулярных взаимодействий, не удовлетворяют точности, необходимой при учете сольватации. Отсутствие достаточно надежных потенциалов делало невозможным использование такого подхода для изучения сольватации. Существенный прогресс в этой области был достигнут благодаря работам Клементи с сотрудниками. Ими была выдвинута идея использовать неэмпирические квантовохимические расчеты для определения потенциалов межмолекулярных взаимодействий [106—109]. Первоначально потенциалы генерировались в численной форме путем неэмпирического расчета энергии взаимодействия сольватированной молекулы и молекулы растворителя. Но, так как дальнейшее использование численного потенциала для построения сольватационной оболочки затруднительно, была подобрана аналитическая функция для его аппроксимации, которая представляла собой сумму атом-атомных потенциалов. При этом все атомы в молекуле были разбиты на классы в зависимости от того, к каким функциональным группам и в каких положениях в группах находится данный атом. В результате число классов во много раз превышало число различных атомов. Для атомов каждого класса подбирались свои атом-атомные потенциалы. Аналитическая форма, в которой производился поиск атом-атомных потенциалов, выбиралась разной и зависела от базиса, использованного в расчете. При вычислении потенциала взаимодействия между молекулами в небольших базисах обычно использовалась относительно простая аналитическая функция
Uij = -Aij/r6ij + Bij/r12ij + Cijqiqj/rij
где Uij - энергия взаимодействия между атомами i и j; rij - расстояние между этими атомами; qi и qj - заряды на атомах; Аij, Вij и Сij - эмпирические параметры, зависящие от того, к каким классам принадлежат атомы i и j.
Для нахождения потенциалов межмолекулярных взаимодействий неэмпирическими методами с использованием больших базисов, близких к хартри-фоковскому пределу, использовались более сложные аналитические функции. Расчеты в больших базисах были проведены для определения аналитических потенциалов, описывающих взаимодействие между молекулами воды. Расчеты в минимальном базисе были использованы для определения атом-атомных потенциалов, описывающих взаимодействие между молекулой воды и основаниями ДНК, аминокислотами и т.д.
Число классов атомов у молекул типа оснований ДНК и аминокислот составляет несколько десятков, число неизвестных параметров в атом-атомных потенциалах достигает нескольких сотен. При определении значений этих параметров приходится варьировать относительное положение и взаимную ориентацию молекул в достаточно широких пределах, практически для вычисления каждого параметра приходится делать 15 - 20 расчетов. Таким образом, для расчета потенциала межмолекулярного взаимодействия молекул среднего размера типа оснований ДНК и молекул воды необходимо сделать несколько десятков тысяч расчетов полной энергии системы неэмпирическим методом. Поэтому процедура подбора параметров связана с очень большими затратами машинного времени. Но следует отметить одно благоприятное обстоятельство: по мере накопления наборов параметров и создания их банка для каждого нового соединения объем вычислений сокращается, так как оказывается возможным отнести большинство атомов к уже известным классам, для которых все параметры аналитических потенциалов известны из расчетов других молекул [110].
После нахождения потенциалов расчет строения сольватационной оболочки и энергии взаимодействия между растворителем и растворенным соединением становится относительно простой задачей, аналогичной задачам конформационного анализа. Аналитическая форма, в которой в настоящее время найдены потенциалы для описания взаимодействия молекул среднего размера и молекул воды, также совпадает с наиболее широко распространенными потенциалами, которые используются в конформационных расчетах. Однако параметры в потенциалах Клементи для межмолекулярных взаимодействий имеют совершенно иную природу. В конформационном анализе потенциалы типа 6 - 12 описывают ван-дер-ваальсовы взаимодействия между атомами, а у Клементи - электронодонорные и электроноакцепторные взаимодействия. Третий член в формуле для атом-атомных потенциалов соответствует кулоновскому взаимодействию. Для электрически нейтральных молекул значение коэффициента Сij в потенциалах Клементи близко к единице. Однако для ионов оно не превышает 0,5; это связано, по-видимому, с эффектами экранирования и перераспределения заряда.
Использование потенциалов Клементи позволяет рассматривать гидратацию весьма сложных молекул большим числом молекул воды. Пока число молекул воды не превышает 10 - 15, каких-либо существенных трудностей при расчете строения гидратационной оболочки не возникает. Однако при дальнейшем увеличении числа молекул воды появляется ряд новых проблем. Для достаточно точного описания гидратационной оболочки даже небольшого соединения количество молекул воды желательно увеличить до 200 - 300. При расчете строения такой огромной гидратационной оболочки основная трудность заключается в существовании большого числа структур с близкими энергиями. Задача сводится к нахождению всех таких структур, определению вероятности реализации каждой из них и усреднению по всем найденным структурам. В таком расчете приходится учитывать температурную зависимость.
В работах Клементи показано, что для нахождения строения гидратационных оболочек можно успешно использовать метод Монте-Карло, с помощью которого были проведены расчеты строения гидратационных оболочек ряда простых ионов с учетом их взаимодействия с 200 - 250 молекулами воды. При этом возникла еще одна проблема. Распределение молекул воды в расчетах методом Монте-Карло носит вероятностный характер, поэтому перед исследователями встала задача перехода к таким простым и наглядным характеристикам гидратации, как число молекул воды в первой гидратационной сфере и ее радиус. Для получения этой информации было предложено вычислить зависимость плотности атомов водорода или кислорода от расстояния до центра иона. На таких графиках получается ряд четко выраженных максимумов. Их положение для атомов кислорода обычно связывают с радиусами гидратационных оболочек, а площадь под кривыми - с количеством молекул воды в оболочке. Ниже приведены радиусы первых гидратационных оболочек (R) и число молекул воды в них (N), вычисленные таким способом [108, 109].
| Ион | R, нм | N |
| Li+ | 0,19—0,20 | |
| Na+ | 0,23—0,24 | 5—6 |
| К+ | 0,28—0,29 | 5—7 |
| F- | 0,27—0,28 | 4—6 |
| Cl- | 0,34—0,35 | 6—7 |
Использование атом-атомных потенциалов весьма перспективно и может существенно расширить наши представления о сольватации и ее влиянии на реакционную способность органических соединений. Банк параметров в настоящее время достаточно велик, и можно надеяться, что в будущем он будет еще расширен. Однако следует подчеркнуть те допущения, которые делаются в этих расчетах.
1. Используется приближенный квантовохимический метод (для молекул среднего размера весьма грубый) для вычисления параметров атом-атомных потенциалов. В случае небольшого числа молекул растворителя ошибки могут быть невелики, но по мере увеличения их количества они будут накапливаться.
2. Ошибки в расчетах могут возникать за счет аппроксимации численного потенциала весьма простыми аналитическими функциями.
3. Атом-атомные потенциалы, которые обычно используют для изучения сольватации, не являются аддитивными функциями, а взаимодействия трех тел учесть довольно сложно и этого почти никогда не делают, хотя эти коллективные взаимодействия существенно влияют на результаты расчета (это, по-видимому, самый большой недостаток метода Клементи).
В ряде работ подход Клементи к учету сольватации был использован для изучения влияния растворителя на поверхности потенциальной энергии органических
Дата добавления: 2020-12-11; просмотров: 567;
Поиск по сайту
Узнать еще
- II Классификация САSЕ-средств
- II. Классификация документов
- II. Формализация процесса формирования математических моделей
- II.4. Классификация нефтей и газов по их химическим и физическим свойствам
- III. Классификация методов воспитания.
- III.1.3. ПРИЧИНЫ НАРУШЕНИЙ СЛУХА. ПСИХОЛОГО-ПЕДАГОГИЧЕСКАЯ КЛАССИФИКАЦИЯ НАРУШЕНИЙ СЛУХОВОЙ ФУНКЦИИ У ДЕТЕЙ
- III.2.3. ПРИЧИНЫ НАРУШЕНИЙ ЗРЕНИЯ. КЛАССИФИКАЦИЯ НАРУШЕНИЙ ЗРИТЕЛЬНОЙ ФУНКЦИИ У ДЕТЕЙ
- VI.2. Классификация месторождений нефти и газа

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
