ДИФФЕРЕНЦИРОВАННЫЕ ФОРМЫ ОЛИГОФРЕНИИ
Хотя описано довольно много синдромов, проявляющихся умственной отсталостью, симптоматика их довольно сходна, так что точный диагноз удается поставить лишь в части случаев.
Хромосомные аберрации являются одной из частых причин олигофре-ний, они составляют 10-12% всех генетически обусловленных задержек психического развития. При этом дефекты наиболее крупных хромосом (с 1-й по 12-ю пары) приводят к нежизнеспособности эмбриона и выкидышам. Трисомии 13-18-й пар хромосом обычно приводят к смерти ребенка на первом году жизни. Другие известные дефекты хромосом - делеция короткого плеча хромосомы 5 (синдром кошачьего крика) и делеция длинного плеча хромосомы 18 (синдром Лежена). Эти синдромы проявляются грубым недоразвитием интеллекта (идиотией или тяжелой имбецильностью). У взрослых чаще встречаются трисомия хромосомы 21 (болезнь Дауна) и дефекты половых хромосом.
Болезнь Дауна - самый частый из хромосомных дефектов. Вероятность рождения больного ребенка составляет 1 случай на 700 новорожденных. Большая часть случаев обусловлена нерасхождением хромосом при образовании яйцеклетки. Такой тип заболевания не связан с патологической наследственностью, зато его вероятность существенно повышается с увеличением возраста матери во время рождения ребенка (у матерей в возрасте 40 лет вероятность в 14 раз больше, чем у 30-летних). Изредка встречаются варианты заболевания, связанные с дислокацией участка хромосомы 21 на нормальную хромосому. В этом случае можно говорить о скрытом носительстве заболевания у родителей, и возраст матери существенной роли не играет, возможны случаи се-
мейного наследования. Еще реже встречается мозаицизм, обусловленный нарушением расхождения хромосом на ранних этапах деления зиготы. Выраженность интеллектуального дефекта в этом случае, как правило, существенно меньше.
Типичные внешние признаки болезни Дауна: небольшой рост, короткие конечности при относительно длинном туловище, широкое, круглое лицо, косой разрез глаз с характерной монголоидной складкой во внутреннем углу глаза (эпикант), деформированные маленькие уши, депигментация радужных оболочек глаз, утолщенный, складчатый, не помещающийся во рту язык, недоразвитые половые органы, общая гипотония мышц и разболтанность суставов. Характерное строение имеет кисть ребенка: пухлая, с укороченными пальцами, искривленным мизинцем и единой поперечной («обезьяньей») складкой. Присутствуют снижение защитных сил организма, распространенный кариес, высокая подверженность инфекциям, пониженная жизнеспособность (часто смерть наступает до достижения полового созревания). Интеллектуальный дефект в большинстве случаев достаточно тяжелый: в 75% случаев - имбецильность, в 20% - идиотия, в 5% - дебильность. Такие дети довольно возбудимы, капризны, речь развита слабо, однако иногда у них можно наблюдать достаточно яркие эмоции. Обычно они привязаны к родителям, стараются им подражать. Характерна ранняя инволюция больных (до 40 лет). В большинстве случаев это сопровождается нарастанием интеллектуальной беспомощности, в мозге отмечают изменения, характерные для болезни Альцгеймера.
Нарушения в распределении половых хромосом приводят обычно к менее грубому интеллектуальному дефекту, чем при болезни Дауна. Трисомия по Х-хромосоме (синдром трипло-Х, генотип XXX) и отсутствие одной Х-хромосомы (синдром Шерешевского-Тернера, генотип Х0) фенотипически проявляются женским полом. И в том и в другом случае отмечают заметные признаки нарушения полового развития (чаще при синдроме Шерешевского-Тернера). Интеллектуальный дефект негрубый (дебильность) и присутствует не у всех пациентов. Фенотипиче-ски синдром Шерешевского-Тернера характеризуется низким ростом, короткой шеей с низкой границей роста волос, наличием крыловидной складки шеи, идущей от сосцевидного отростка височной кости к лопатке, недоразвитием вторичных половых признаков и первичной аменореей. Эти пациенты довольно добродушны, трудолюбивы, послушны. При трисомии по Х-хромосоме снижение интеллекта отмечают в 75% случаев, однако оно негрубое, поведение психопатоподобное. Нередко возникают психозы, напоминающие шизофрению.
Генотипы XXY и XYY выявляют у мужчин. Синдром Кляйнфел-тера выражается присутствием лишней Х-хромосомы при наличии Y-хромосом (встречаются варианты XXY, XXXY, XXYY). Пациенты с такой патологией характеризуются инфантилизмом, гипогонадизмом, евнухоидным телосложением, гинекомастией, бесплодием. Интеллектуальный дефект обнаруживают часто, но не всегда. Характерны мягкость, ранимость, психический инфантилизм, иногда стеснительность и переживания по поводу своего дефекта. Наличие лишней Y-хромосомы (синдром XYY) обычно не проявляется грубыми соматическими расстройствами. У таких мужчин не нарушена половая функция, они могут иметь нормальных детей, без генетических дефектов. Психическое недоразвитие отмечают у 80% пациентов, оно обычно бывает негрубым (легкая дебильность). Такие мальчики с детства отличаются высоким ростом, эмоциональной неустойчивостью, высокой частотой взрывных реакций, склонностью к правонарушениям.
Наследуемые генные дефекты обычно проявляются недостаточностью того или иного фермента. Во внутриутробном периоде данный дефект компенсируется за счет организма матери, поэтому в большинстве случаев при рождении не отмечают заметного отличия от нормы. Однако при длительно существующей ферментной недостаточности энзимопатия может приводить к довольно тяжелой олигофрении. Эн-зимопатии составляют от 5 до 10% всех случаев тяжелой умственной отсталости. В большинстве случаев данные заболевания обусловлены рецессивным геном (табл. 26.2). Проявления олигофрений, вызванных рецессивным геном, часто более тяжелые, чем передаваемые по доминантному типу, поскольку доминантный ген чаще встречается в гетерозиготном состоянии, и имеющийся здоровый ген может частично компенсировать дефект.
Таблица 26.2. Примеры синдромов наследственных форм олигофрений

Окончание табл. 26.2
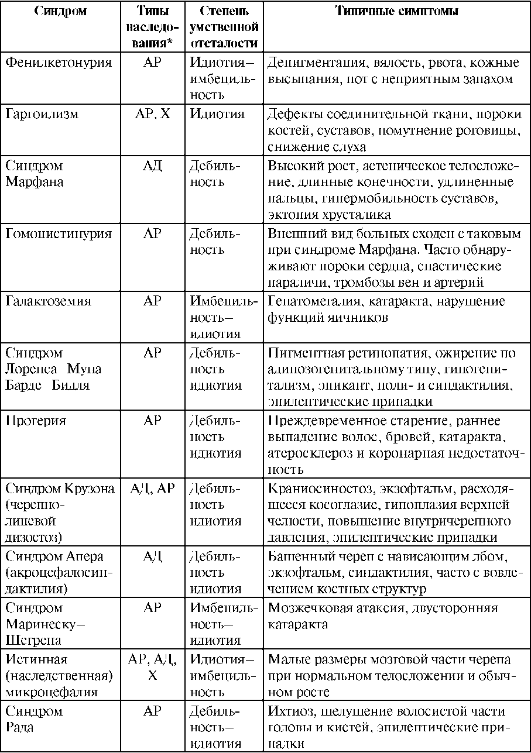
Синдром Мартина-Белл (Fragile X Syndrome) - сцепленный с полом генный дефект Х-хромосомы, который приводит к образованию на ней сегментов и перетяжек. После разработки методик определения данного дефекта оказалось, что он встречается чрезвычайно часто (1 на 1500 мальчиков и 1 на 2500 девочек) и является второй по частоте после болезни Дауна изученной причиной умственной отсталости. Спектр психических расстройств весьма широк - от аутизма до рас-торможенности и неуправляемости. Интеллектуальный дефект колеблется между дебильностью и имбецильностью. Характерна скачущая речь с большим количеством повторов. Во внешности обращают на себя внимание большая голова, широкий лоб, особая форма носа, оттопыренные уши. Матери больных мальчиков бывают носительницами патологического гена (30% из них имеют признаки заболевания). Загадкой остается, почему 20% мужчин, наверняка получивших данный ген от матери, остаются здоровыми. Методов лечения нет.
Фенилкетонурию описал А. Феллинг в 1934 г. Ее регистрируют в 1 случае на 10 000 новорожденных, с одинаковой частотой у мальчиков и девочек. Поскольку болезнь передается по аутосомно-рецессив-ному типу, оба родителя ребенка являются здоровыми носителями, и вероятность рождения второго ребенка с той же патологией составляет 25%. Распространенность здоровых носителей данного гена в популяции составляет 1:50. Проявления заболевания обусловлены отсутствием фермента фенилаланингидроксилазы. Это вызывает нарушения превращения фенилаланина в тирозин, который, в свою очередь, служит важнейшим предшественником нейромедиаторов (норадре-налина, дофамина), гормонов (адреналина, тироксина) и меланина. Хотя при рождении дети ничем не отличаются от здоровых, однако уже в первые месяцы отмечают нарастание вялости, заторможенности, слабую реакцию на окружающее, замедление психического развития. Недостаток меланина проявляется светлыми волосами и голубым цветом глаз. Довольно часто возникает рвота, особенно плохо дети переносят введение прикорма. Часто наблюдают кожные высыпания, повышенная потливость со специфическим, неприятным запахом пота. При отсутствии диетического лечения, начатого на первом году жизни (лучше не позже 2-3 мес), развивается тяжелая олигофрения (в 65% случаев - идиотия). Для своевременной диагностики двукратно исследуют мочу ребенка в возрасте до 2-3 мес. Применяют реакции с полуторахлори-стым железом (пробу Феллинга), с динитрофенилгидразином и микробиологический тест Гатри. Однако эти тесты неспецифичны, поэтому
окончательная диагностика основана на определении содержания фе-нилаланина в сыворотке крови. Единственным методом лечения служит строгая диета с ограничением белков растительного и животного происхождения (мяса, яиц, рыбы, сыра, бобовых). Недостаток незаменимых аминокислот компенсируют специально приготовленными смесями. Из продуктов допустимо употребление блюд из овощей, фруктов, сахара, меда, картофельного крахмала, жиров.
Гаргоилизм - группа синдромов, проявляющихся накоплением в организме кислых мукополисахаридов. Эти вещества имеют большое значение для развития соединительной ткани, поэтому выявляются множественные дефекты костей, суставов, позвоночника, черепа, пороки сердца, грыжи, изменения в органах зрения. Находят также изменения мозга, гидроцефалию, утолщение твердой мозговой оболочки. Заболевание обусловлено рецессивным геном, в части случаев регистрируют сцепленное с полом наследование (соотношение мальчиков и девочек составляет 2:1). Признаки заболевания проявляются в первые месяцы жизни и быстро нарастают, в типичных случаях достигая степени идиотии. Ранняя диагностика возможна путем исследования мочи на кислые мукополисахариды (реакции Барри и Дорфмана). Специфической терапии не найдено. Применяют АКТГ и гормоны щитовидной железы. Некоторый эффект может быть достигнут при использовании высоких доз ретинола (витамина А) и рентгеновском облучении гипофиза.
Другие заболевания, передаваемые по аутосомно-рецессивному типу: гомоцистинурия, лейциноз (болезнь кленового сиропа, синдром Мен-кеса), галактоземия, фруктозурия и сукрозурия. Гомоцистинурия (нарушение метаболизма метионина) проявляется негрубым интеллектуальным дефектом и значительными изменениями в опорно-двигательном аппарате, строении глаз. Нередко отмечают спастические параличи и тромбозы. Нарушения в усвоении углеводов (галактозы, фруктозы и сукрозы) проявляются диареей, гипотрофией, задержкой психического развития и могут стать причиной смерти в детском возрасте. Лейциноз (болезнь кленового сиропа) - злокачественно протекающее заболевание, приводящее к смерти на 1-2-м году жизни, обусловленное нарушением обмена сразу трех аминокислот (лейцина, изолейцина и валина).
Синдром Марфана (арахнодактилия) передается по аутосомно-доми-нантному типу и часто наблюдается у нескольких членов одной семьи. Интеллектуальный дефект обычно негрубый, иногда отсутствует. У па-
циентов отмечают высокий рост, тонкие длинные руки и пальцы, гипермобильность суставов, астеническое телосложение. В психическом отношении характерны замедленность движений, тугоподвижность психических процессов, отсутствие инициативы. Отмечают плохую устойчивость к инфекциям, что иногда становится причиной ранней смерти.
При внутриутробных инфекциях и интоксикациях выраженность повреждений зависит не только от характера воздействия, но и от временного периода, в котором такое воздействие было оказано. Особенно тяжелые нарушения развития, сопровождаемые грубыми дефектами органов, возникают при инфекциях и интоксикациях, действовавших в I триместре беременности. Практически любая инфекция у беременной в этот период может стать причиной поражения эмбриона. Чаще других встречаются поражения, вызванные вирусами краснухи и гриппа. Сифилис, токсоплазмоз, листериоз, цитомегалию выявляют значительно реже.
Поражение вирусом краснухи в I триместре беременности приводит к порокам развития в 12% и мертворождению - в 7,2% случаев (при инфицировании во II триместре беременности эти показатели в 2-3 раза меньше). В европейских странах краснуха возникает редко (наиболее тяжелые эпидемии зарегистрированы в Австралии). При рубеолярной эмбриопатии олигофрения нередко сочетается с пороками развития различных органов: глаз, сердечно-сосудистой системы, слухового аппарата, скелета и зубов, мочеполовых органов. Психическое недоразвитие часто очень глубокое. Для профилактики краснухи беременных проводят вакцинацию до наступления беременности.
Тяжелая олигофрения характерна и для других внутриутробных инфекций: листериоза, токсоплазмоза, сифилиса, цитомегалии.
Листерия - широко распространенный грамположительный микроорганизм, выделяемый у многих домашних животных, которые обычно и становятся источником заражения. При внутриутробном заражении возникают детский септический гранулематоз и менингоэнцефалит. В остром периоде возможно лечение антибиотиками и сульфаниламидами. Изредка встречаются и постнатальные формы листериоза с поражением нервной системы.
Токсоплазмоз - паразитарная инвазия. Считают, что от 10 до 30% населения в разных странах - здоровые носители Toxoplasma gondii, развитие заболевания связывают с особенностями иммунного статуса человека. Отмечают более высокий риск возникновения шизофрении у людей с высоким титром антител к этому паразиту. Ранний
внутриутробный токсоплазмоз чаще заканчивается мертворождением. Более позднее и постнатальное заражение бывает причиной олигофрении. Клинические проявления такой олигофрении неспецифичны и во многом зависят от времени заражения. Чаще других расстройств регистрируют хориоретинит, микрофтальмию, катаракту, внутримоз-говые обызвествления, гидроцефалию, микроцефалию, часто эпилептические припадки. Диагностика основана на обнаружении паразитов в СМЖ. Ориентировочный диагноз может быть подтвержден положительной кожной аллергической реакцией. Для лечения внутриутробной инфекции используют спирамицин (ровамицин*), при постнатальном заражении - пириметамин + сульфадоксин (фансидар*).
Характерные симптомы врожденного сифилиса: гнойно-кровянистые выделения из носа, увеличение поверхностных лимфатических узлов, мокнущие кожные высыпания. Часто отмечают множественные дефекты развития: деформации черепа, седловидный нос, утолщение и искривление костей (саблевидные голени), триаду Гетчинсона (полулунные выемки на верхних резцах, кератит и поражение среднего уха). В неврологическом статусе обнаруживают характерные для сифилиса зрачковые симптомы (симптом Аргайла Робертсона), параличи, парезы, тики, хореиформные гиперкинезы и судорожные припадки. Для того чтобы предотвратить прогрессирование заболевания, необходимы своевременная серологическая диагностика (реакции Вассермана и пассивной гемагглютинации) и специфическое лечение (антибиотиками, препаратами йода и висмута).
Цитомегаловирусная инфекция считается довольно распространенной, однако у большинства людей носительство протекает бессимптомно (в одном из исследований патология обнаружена у 2 из 26 инфицированных детей). Характерны микроцефалия, поражение глаз, зрительного и слухового нервов. У части детей нарушения возникают не при рождении, а позже. Лабораторная диагностика возможна только в первые месяцы после рождения, химиотерапия неэффективна.
Гемолитическая болезнь новорожденных приводит к тяжелой интоксикации и формированию билирубиновой энцефалопатии. Частота гемолитической болезни составляет 1 случай на 250-300 родов, ее причина - несовместимость матери и плода по резус-системе, реже по системе АВ0. Резус-конфликт возникает в результате иммунизации резус-отрицательной беременной резус-положительной кровью плода. Вероятность такой иммунизации составляет около 10% после первой и каждой последующей беременности, включая те, что закончились
абортом (если мать беременной также была резус-отрицательной, то риск повышается). Для профилактики иммунизации проводят внутримышечное введение иммуноглобулина человека антирезус Rho (D) резус-отрицательной матери начиная с 28-й недели беременности (но не позднее 72 ч после родов). Гемолиз крови ребенка сопровождается накоплением непрямого билирубина, что нарушает работу дыхательных ферментов. Особенно страдают липофильные ткани (базальные ганглии, кора головного мозга, надпочечники). Олигофрения в результате гемолитической болезни нередко сопровождается экстрапирамидными двигательными расстройствами и дефектами слуха. Интеллектуальный дефект зависит от тяжести заболевания и может колебаться от дебиль-ности до тяжелой идиотии. Нарушениям слуха сопутствуют грубые расстройства речи. Характерны возбудимость, назойливость, раздражительность, резкое снижение критики и эйфория. Единственным методом терапии служит раннее обменное переливание крови в первые сутки жизни. Также назначают магния сульфат внутрь и глюкокортико-иды в постепенно снижаемых дозах.
Осложнения при родах включают асфиксию, кровоизлияния и травмы. Разделить эти причины не всегда возможно, поскольку нередко они возникают одновременно. Картина олигофрении при этом не отличается специфичностью, конкретные симптомы во многом зависят от локализации и тяжести поражения. Соответственно степень олигофрении может быть различной - от легкой дебильности до тяжелой идиотии. Чаще, чем при наследственных формах олигофрении, возникает нарушение таких предпосылок интеллекта, как память и внимание. Весьма вероятно возникновение неврологической симптоматики - парезов и параличей, псевдобульбарной дизартрии, алалии, судорожных припадков. Признаки олигофрении сочетаются с такими проявлениями психоорганического синдрома, как истощаемость и утомляемость. Нередко обнаруживают внутричерепную гипертензию.
В постнатальном периоде важнейшей причиной олигофрении становятся тяжелые мозговые инфекции (менингиты и менингоэнцефалиты). Во многом тяжесть последствий инфекции зависит от индивидуальной реактивности организма. Иногда такие инфекционные заболевания, как корь, ветряная оспа, коклюш, пневмония, возникая в раннем детском возрасте, вызывают вторичный энцефалит, который становится причиной умственной отсталости.
Травмы у детей первых лет жизни редко становятся причиной задержки психического развития, поскольку в этом возрасте дети нахо-
дятся под постоянным наблюдением родителей. Клиническая картина посттравматических олигофрений мозаична, не все психические функции нарушаются в одинаковой степени, что делает такую олигофрению более сходной с органическими деменциями, возникающими в более старшем возрасте. В постнатальном периоде также возникают эндокринопатии. Чаще других встречается задержка развития вследствие недостаточности щитовидной железы - кретинизм. Отмечают как редкие наследственные формы заболевания, так и варианты, вызванные экзогенными вредностями (недостатком йода, аутоиммунным или инфекционным поражением щитовидной железы и гипофиза). Заболевание значительно чаще развивается у девочек. Характерны малый рост, нарушение развития зубов, замедление окостенения, артериальная гипотензия и брадикардия, атония кишечника. В ряде случаев своевременное назначение гормонов щитовидной железы (левотироксина натрия, лиотиронина) предупреждает развитие тяжелого психического дефекта.
Дата добавления: 2022-02-05; просмотров: 648;
Поиск по сайту
Узнать еще
- III. Суммарные допуски формы расположения
- V. Сборка и нагружение формы.
- V.VII. Зависимость ширины и формы выхода слоя на поверхности от его истинной мощности, угла падения и формы рельефа
- А - с прямолинейной спинкой; б - с криволинейной спинкой; в - с канавкой удлиненной формы
- А) Контроль отклонений от правильной цилиндрической формы.
- А. Локализованные формы
- Автоматический контроль точности размеров и формы деталей. Разновидности контроля. Использование информации, полученной при контроле,
- Аккумулятивные и абразионные формы рельефа побережья.

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
