Методы расчетов электронной структуры
Наиболее удобным, с точки зрения квантовой химии, является такой метод расчета структуры и свойств молекул, который использовал бы информацию только о конфигурации электронных оболочек атомов, составляющих систему. Реализация такого метода позволила бы исследователям предсказывать существование и свойства новых материалов, еще не полученных в эксперименте, например, сверхтвердых материалов и др.
Ab initio, т. е. первопринципные методы, используют вышеописанный принцип, однако из-за высокой сложности расчета в них применяются некоторые приближения, которые не позволяют применить эти методы к любым системам. При этом точность расчета в большинстве случаев довольно высока.
К основным методам расчета из первых принципов можно отнести следующие:
– метод Хартри – Фока и его дальнейшие развития;
– метод функционала электронной плотности.
Метод Хартри – Фока, или метод самосогласованного поля, является одним из эффективных методов решения задач квантовой химии. Его идея состоит в том, что взаимодействие электрона с его окружением заменяется взаимодействием с неким усредненным полем 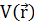 . Таким образом, не решаемая квантовомеханическая задача многих тел сводится к решению одночастичного уравнения
. Таким образом, не решаемая квантовомеханическая задача многих тел сводится к решению одночастичного уравнения 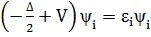 (где Δ – оператор Лапласа).
(где Δ – оператор Лапласа). 
В методе функционала плотности вместо волновых функций электронов  используется электронная плотность – функция трех пространственных переменных
используется электронная плотность – функция трех пространственных переменных  . Затем находится минимум энергии, являющейся функционалом от электронной плотности. Это позволяет существенно упростить задачу и рассчитывать системы из десятков и сотен электронов.
. Затем находится минимум энергии, являющейся функционалом от электронной плотности. Это позволяет существенно упростить задачу и рассчитывать системы из десятков и сотен электронов.
Решения уравнения Хартри – Фока без использования каких-либо других приближений лежат в основе неэмпирических методов расчета, которые отличаются используемым базисом атомных орбиталей. При использовании этих методов основные затраты машинного времени (около 70 %) направлены на вычисление интегралов межэлектронного взаимодействия оператора Фока. По мере увеличения размеров молекулы число таких интегралов возрастает примерно пропорционально N4, где N – число базисных функций. Соответственно этому растут время и стоимость расчета.
Наиболее перспективными являются методы, основанные на замене большей части интегралов параметрами, вычисленными с помощью данных, взятых из эксперимента (потенциалами ионизации атомов в орбитальных валентных состояниях и др.), и использовании различных приближенных выражений для оценки интегралов. Основанные на этом подходе методы называются полуэмпирическими.
Среди полуэмпирических методов расчета можно выделить следующие:
– метод молекулярных орбиталей Хюккеля;
– расширенный метод Хюккеля;
– метод сильной связи.
Даже после появления таких быстрых алгоритмов, как семейство O(N)–алгоритмов, время счета в которых пропорционально количеству атомов в системе, системы, насчитывающие 104 – 106 атомов, невозможно просчитать на уровне решения уравнения Шредингера или его аналогов (уравнения Хартри – Фока, Кона – Шэма) для волновых функций электрона. Решение таких уравнений требует проведения огромного количества вычислений для матричной диагонализации и поэтому практически нереально для систем вышеперечисленных размеров. Современные же технологии, особенно нанотехнологии, фармацевтика и микробиология, требуют возможности рассчитывать системы, насчитывающие десятки тысяч атомов и выше, пусть даже и с минимумом получаемой информации, например – полной энергии системы. При этом часто возникает потребность выполнения молекулярно-динамических расчетов, где поведение системы моделируется в дискретном времени с шагом порядка 1.0 фемтосекунды (10–15 с) и в течение времени, много большего обратной частоты фононных колебаний, обычно 10–6 – 10–9 с, что на несколько порядков замедляет проведение расчетов. Зато при этом моделируется поведение системы при любой заданной температуре с минимальными погрешностями.
Из вышесказанного следует, что огромные системы практически невозможно просчитать на уровне вычисления волновых функций электронов, и поэтому для них применяются методы, базирующиеся на эмпирическом знании потенциалов взаимодействия атомов или ионов, без вычисления электронных волновых функций или плотностей.
Основными эмпирическими методами расчета электронной структуры являются:
– метод парных потенциалов;
– метод многочастичных потенциалов;
– метод молекулярной динамики.
Дата добавления: 2016-05-31; просмотров: 2117;
Поиск по сайту
Узнать еще
- I. История открытия и методы исследования вирусов
- ID_структуры . ID_поля
- II. Категории и методы политологии.
- III. Методы искусственной физико-химической детоксикации.
- А) Назначение и порядок выполнения расчетов
- Абсолютный возраст горных пород и методы его определения
- Автоматизированная информационная система организации перевозок грузов по безбумажной технологии с использованием электронной накладной (АИС ЭДВ)
- Автоматические методы изготовления фотошаблонов.

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
