И ЧАСТОТЫ ВНУТРИМОЛЕКУЛЯРНЫХ КОЛЕБАНИЙ
Силовая постоянная химической связи K(i), равная второй производной полной энергии молекулы по удлинению данной связи qi, (∂2E/∂qi 2), является, пожалуй, основной характеристикой механических свойств связи. Для одноименных связей в разных молекулах сходного типа она варьируется в небольших пределах. Известны полуколичественные корреляции ее с длиной и порядком связи, а также некоторые взаимосвязи с энергией диссоциации.
Химическая связь в многоатомной молекуле, строго говоря, не может быть охарактеризована одним силовым коэффициентом. Для этой цели требуется введение матрицы коэффициентов, включая те, которые связаны с взаимодействием данной связи и всех других связей, т.е. недиагональные элементы [132,133]: K(i,j) = ∂2E/∂qi ∂gj. Последние определяют появление сил, действующих в j-x связях, в результате растяжения i-й связи. Недиагональные элементы, как правило, невелики, но для решения некоторых задач они имеют первостепенное значение.
Если располагать данными о структурной формуле соединения и необходимым набором силовых постоянных, то для изучаемой конформации можно довольно просто вычислить частоты всех нормальных колебаний атомов ω, которые проявляются в ИК- и КР-спектрах, электронно-колебательных спектрах поглощения и вторичного излучения. Можно также определить формы нормальных колебаний и охарактеризовать силовое поле в как угодно деформированной молекуле. Эти возможности лимитируются недостаточностью сведений о самих силовых постоянных, которые особенно ограниченны для электронно-возбужденных состояний.
Общее количество информации, требующейся для точного расчета частот ω сложных молекул, весьма велико, и имеющиеся отрывочные данные приходится дополнять, используя аналогии, а иногда и весьма сомнительные допущения. В этих условиях привлечение дополнительных данных представляется очень важным. Квантовая химия занимает здесь особое место, так как позволяет в принципе не только оценить любые силовые постоянные как для основного, так и для электронно-возбужденных состояний, но и выяснить для отдельных коэффициентов их происхождение и связь со строением электронной оболочки, чему, к сожалению, пока не уделялось достаточного внимания.
Известно, что решение обратной задачи, т.е. вычисление силовых коэффициентов по известным из эксперимента колебательным частотам, требует большой и трудной подготовительной работы по синтезу изотопных моделей. Во многих случаях решение неоднозначно и выбор между наборами коэффициентов, удовлетворяющих исходной системе уравнений, основывается нередко на интуиции. Привлечение методов квантовой химии дает в распоряжение исследователя независимые критерии для такого выбора. К сказанному можно добавить, что квантовохимический расчет силовых постоянных не связан с какими-либо дополнительными трудностями и может быть выполнен обычными методами, описанными во введении.
Для расчета силовых постоянных довольно широко применяют как полуэмпирические, так и неэмпирические методы квантовой химии. В любом случае сначала оптимизируют геометрию, т.е. определяют наиболее устойчивую конформацию, отвечающую минимуму полной энергии; затем вычисляют вторые производные полной энергии по декартовым или естественным координатам (силовые постоянные), а при необходимости - кубичные и биквадратичные члены.
Выбор базиса существенно сказывается на результатах неэмпирических расчетов силовых постоянных (в особенности недиагональных элементов). Так, при использовании минимального слейтеровского базиса согласие с экспериментом получается весьма посредственное. Точность расчета существенно повышается при переходе к валентно-расщепленному базису (4-31ГФ, 4-21ГФ или 3-21ГФ), но его дальнейшее расширение уже малоэффективно. При выходе на так называемый хартри-фоковский предел силовые постоянные оказываются завышенными на 10 - 20% [134], при этом точность расчета недиагональных элементов намного ниже. Так, для молекулы СО2 значение К(С—О, С—О/) получилось завышенным почти в 2 раза [135]. Учет электронной корреляции улучшает результаты [136, 137], однако для многоатомных молекул он практически недоступен.
Для полуэмпирических методов характерно относительное занижение частот деформационных колебаний по сравнению с валентными. При этом в методе ППДП/2 без введения поправок значения колебательных частот для деформационных мод хорошо согласуются с экспериментом, а данные для валентных колебаний получаются сильно завышенными. В методе МЧПДП последние, напротив, близки к опытным данным, но частоты деформационных колебаний занижены. Методы МЧПДП/3 и МПДП дают результаты промежуточного типа: значения частот для валентных колебаний получаются немного завышенными, а для деформационных - заниженными. Ошибки при использовании методов МЧПДП/3 и МПДП получаются примерно такие же, как в неэмпирических расчетах без учета электронной корреляции (в среднем около 200 см-1).
В связи с тем, что ошибки в большинстве случаев носят систематический характер, их удается значительно уменьшить введением эмпирически подобранных масштабных корректирующих множителей для определенных типов силовых постоянных (например, для связей С—С, углов С—С—Н и т.д.) или инкрементов, которые прибавляются к рассчитанным частотам.
Для расчетов методами МЧПДП/3 и МПДП Дьюар и Форд [138, 139] подобрали систему инкрементов, специфичных для валентных, деформационных и торсионных колебаний определенных атомных групп или связей; на очень большом числе примеров продемонстрирована удовлетворительная точность результатов. Однако многообразие форм нормальных колебаний, которые в сложной молекуле включают колебания многих связей и углов, и невозможность четкого разграничения валентных и деформационных мод ограничивают перспективы корректирования частот по типам колебаний.
Более логичным представляется корректирование значений силовых постоянных, и на этом пути достигнуты положительные результаты. В настоящее время используется несколько методик подбора корректирующих множителей. Они различаются способом корректировки недиагональных силовых коэффициентов. В' работе [140] предложено корректировать только диагональные силовые постоянные, а недиагональные оставить без изменений. Этот подход использован в работе [141], в которой методом МЧПДП/3 рассчитаны частоты нормальных колебаний этана, пропана, этилена, бутена-2 и 1,3-бутадиена. Для улучшения количественного согласия с экспериментом были уменьшены силовые постоянные валентных связей на 30%, увеличены силовые постоянные валентных и торсионных углов на 20%, а недиагональные коэффициенты оставлены без изменений. Частоты нормальных колебаний исследованных соединений, вычисленные с такой корректировкой силового поля, отличаются от экспериментальных значений в среднем на 50см-1.
Блом и Алтона [142 - 144] использовали один общий корректирующий множитель для всех недиагональных силовых постоянных. Так, при неэмпирическом расчете в базисе 4-31ГФ колебательных спектров этилена ими использовано пять корректирующих множителей порядка 0,75 - 1,0. Из них четыре - для диагональных силовых постоянных и один - для всех недиагональных членов. При этом среднее расхождение с наблюдаемыми на опыте 12 колебательными частотами уменьшилось более чем в 10 раз и составило около 7 см-1 [142]. Подобные результаты получены и для этана, пропана, пропена и диметилового эфира, но корректирующие коэффициенты подбирались для каждого соединения индивидуально [143, 144]. Отметим, что простое определение частот по набору коэффициентов, полученных из опыта для других молекул сходного типа, дает при отсутствии сопряжения примерно такую же точность.
В работах Пулея с сотрудниками [145, 146] предложено подбирать значения корректирующих множителей только для диагональных членов, а корректирующие множители для недиагональных коэффициентов вычислять как среднее геометрическое из соответствующих диагональных величин, т.е. если Сi и Сj - корректирующие множители для K(i) и K(j), то корректирующий множитель для K(i,j) - 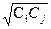 . В настоящее время методика Пулея используется наиболее широко.
. В настоящее время методика Пулея используется наиболее широко.
Первые работы с использованием корректирования силового поля молекулы по методике Пулея выполнены полуэмпирическими методами. Так, было показано [146], что при расчете колебательных частот углеводородов методом ППДП/2 следует ввести следующие корректирующие множители: 0,38 для силовых постоянных валентных связей; 0,80 для силовых постоянных валентных углов; 0,62 для силовых постоянных углов выхода из плоскости молекулы. Такие же поправки были использованы для глиоксаля и акролеина [147]. Набор масштабных множителей для органических соединений, содержащих атомы N и О, рекомендован и опробован в работе [148], при этом среднеквадратичная ошибка в ряду весьма сложных триазолов не превышала 30 см-1.
Были рассчитаны [149] частоты нормальных колебаний 4Н-пиран-4-она методами ППДП/2 и МЧПДП/3. Для каждого метода подобран свой набор корректирующих множителей; при этом предполагалось, что их значения для связей и углов, образованных разными атомами, различаются. В случае метода ППДП/2 значения вычисленных корректирующих множителей для валентных связей лежат в области 0,25 - 0,45; для валентных углов в плоскости молекулы — 0,83 - 1,20; для углов выхода из плоскости молекулы — 0,65 - 0,80. При использовании метода МЧПДП/3 значения подобранных корректирующих множителей для валентных связей лежат в области 0,70 - 0,80; для валентных углов — 1,15 - 1,30; для углов выхода из плоскости молекулы — 1,20 - 1,30. Средние отклонения рассчитанных колебательных частот от экспериментальных величин в случае метода ППДП/2: для плоских колебаний 20 см-1, для неплоских колебаний 33 см-1; в случае метода МЧПДП/3: для плоских колебаний 25 см-1, для неплоских колебаний 17 см-1.
Опубликовано много неэмпирических расчетов силовых полей и колебательных спектров молекул небольшого и среднего размеров с использованием методики Пулея [145, 150 - 152]. Для малых молекул расхождения между вычисленными и экспериментальными частотами нормальных колебаний составили несколько обратных сантиметров [145, 150]. Для молекул среднего размера это расхождение несколько больше. Так, для 1Н-пиррол-2,5-диона среднеквадратичное отклонение вычисленных частот нормальных колебаний от экспериментальных величин составило 7 см-1 для плоских и 10 см-1 для неплоских мод [151], для имидазола эти величины равны соответственно 9 и 27 см-1 [152].
При подборе масштабных множителей или инкрементов естественно встает вопрос об их воспроизводимости в ряду разных молекул. В табл. 3.1 приведены корректирующие множители для 1Н-пиррол-2,5-диона.
Близкие значения коэффициентов из наборов 1 и 2 демонстрируют хорошую переносимость корректирующих множителей между молекулами, которые не являются родственными, но содержат однотипные валентные связи и углы. Может показаться, что вопрос о переносимости корректирующих множителей сходен с вопросом о переносимости определенных из опыта силовых постоянных, но в действительности происхождение ограничений универсальности в этих двух случаях совершенно различно; в первом случае в основе ограничения лежит существенная иррегулярность ошибок выбранного метода расчета, а во втором - реально существующая специфика силового поля у тех или иных исследуемых молекул, которая сама по себе чрезвычайно интересна и важна для химии и к тому же доступна для изучения квантовохимическими методами.
Таблица 3.1
Дата добавления: 2020-12-11; просмотров: 516;
Поиск по сайту
Узнать еще
- Автогенераторы гармонических колебаний.
- Автоколебательные системы с внешним воздействием, синхронизация колебаний
- Автоматическая подстройка частоты
- Автоматическое ограничение повышения частоты
- Автоматическое ограничение снижения частоты
- Алгебраический метод определения симметричных колебаний
- Алгебраический способ определения симметричных автоколебаний и устойчивости
- Амплитрон — это лампа обратной волны магнетронного типа (ЛОВМ). Предназначен для усиления СВЧ колебаний.

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
