Неопределенность состава соединений в растворах
3агадочностью своей природы растворы постоянно привлекали внимание химиков. Они по разному рассматривались Бертолле и Прустом. Бартолле считал их истинными химическими соединениями, так как не видел принципиальной разницы между природой сил сцепления составных частей в растворах, с одной стороны, и во всех других телах, с другой. Пруст же рассматривал растворы как «совокупности сочетаний, которые не связываются природой прочно» и потому не могут иметь определенных и постоянных пропорций. Однако ни Бертолле, ни Пруст не могли привести убедительных прямых экспериментальных обоснований своим взглядам на растворы. Точка зрения Пруста в первой четверти прошлого столетия приобрела большинство сторонников только потому, что к химическим соединениям стали относиться лишь химические индивиды постоянного состава. Ведь Пруст и сам не отрицал наличие какого-то взаимодействия, а именно определенного притяжения составных частей в растворах. Он только подчеркивал, что этот вид притяжения отличается несравненно более слабыми силами от другого вида, каковым наделены соединения. «Эти два вида притяжения настольно отличны по своим результатам, - говорил он,- что смешивать их никак невозможно» .
Но что же представляют собою эти относительно слабые силы взаимодействия частиц раствора? Где проходит граница между этими «слабыми силами, присущими взаимодействию частиц в растворах, и «пpочной» силой химических связей в соединениях? На эти вопросы в XIX в. различные исследователи отвечали по-разному. Для Пруста, например, было существенным лишь отграничение соединений как «привилегированных продуктов природы», т.е. продуктов, имеющих постоянный состав, от всех других смесей, в том числе и растворов. Для Дальтона этот вопрос также не был существенным, ибо он решался им а priori путем вывода растворов за пределы «химической атомистики». Это было очень странно, ибо Дальтон, подобно Бертолле, рассматривал и химическое взаимодействие и взаимодействие частиц при конденсации газов в жидкие и твердые тела как проявление сил всемирного тяготения. Различие в интенсивности сцепления атомов в сложном атоме, с одной стороны, и сил взаимодействия частиц в смесях и растворах, с другой, было для него достаточным, чтобы не причислять растворы к химическим соединениям. Но каково это различие, т.е. опять-таки где проходит граница между прочными силами сцепления, причисляемыми к химическим, и слабыми, относимыми к механическим силам. Дальтон не указывал и не мог тогда указать.
Берцелиус для отграничения растворов от химических соединений имел несравненно больше оснований. Принимая так же, как и другие исследователи, силы сцепления атомов в химической частице за прочную химическую связь, он считал, что последняя обусловлена не всемирным тяготением, а особым проявлением электричества - наиболее полной взаимной нейтрализации разноименно заряженных атомов и атомных групп. Электрохимической природой сил сродства он объяснял и те способы сочетания атомов, которые приводят к относительно несложной частице постоянного состава, являющейся носительницей стехиометрических отношений. Явления же растворения, согласно Берцелиусу, «не сопровождаются никакой электрохимической нейтрализацией» и поэтому «сила соединения, на которой основано растворение твердого вещества в жидкости, не идентична силе, от действия которой образуются химические соединения, и не должна быть смешиваема с нею». По мнению Берцелиуса, растворение представляет собою механический процесс образования особого рода смесей.
Берцелиус тоже не мог указать в смысле количественной оценки границу между прочными химическими связями (которые мы теперь оцениваем в 70-80-100 ккал/моль) и слабым взаимодействием частиц в растворах (имеющим по современной оценке энергию связи от 5-7 до 20-30 ккал на моль). Но зато он выдвинул другой, качественный, критерий для определения существенных различий между химическим и иными видами взаимодействия. Считая последние проявлением сил всемирного тяготения, для химического взаимодействия атомов в частице он указал иную природу - электричество.
Но уже в первой половине XIX столетия появилось немало ученых, которые не только выступили сторонниками химических концепций в области растворов, но и стремились экспериментально обосновать химическую природу растворения. К таким исследователям относились Г. И. Гесс, Т. Грэм, Х. Поггендорф, Л. Гмелин, Г. Копп, Д. Н. Абашев и другие. Многочисленные работы этих ученых, выяснившие достаточно отчетливо выступающую зависимость изменения объема тел при растворении, с очевидностью указывали не только на взаимодействие компонентов раствора, но и на образование своеобразных химических соединений. В одних случаях при этом появились указания на образование более или менее определенных соединений - гидратов или сольватов, в других - приходилось делать вывод о наличии в растворах неопределенных соединений, т.е. веществ переменного состава. Во всех случаях, однако, исследователи подчеркивают, что силы химического взаимодействия при образовании такого рода соединений относительно слабее тех полновалентных сил, которые связывают атомы в молекуле. «Силы, действием которых образуются растворы, - писал в 1858 г. Д. Н. Абашев,- находятся в чрезвычайно близком соотношении с теми силами, от которых зависит молекулярное состояние тел, кристаллизация, явления капиллярные и т. п.; поэтому растворы могут быть названы соединениями молекулярными». К таким же выводам в результате своих работ приходили Г. Копп, П. Крамерс, Ф. Рудберг и др. Косвенные указания на существование в растворах молекулярных соединений типа сольватов в ряде случаев были подтверждены прямой фиксацией, таких веществ путем выделения их в кристаллическим виде при неоднократной повторяемости опытов.
В 1860-х годах появилось довольно много работ, показывавших, что границы между определенными дальтоновскими соединениями и неопределенными соединениями в растворах не столь уже существенны. А. Сент-Клер-Девилль экспериментально доказал, что в растворе имеет место динамическое равновесие между молекулярными соединениями и продуктами их термической диссоциации - молекулами или ассоциатами, т.е. агрегатами молекул меньшего молекулярного веса. Почти одновременно с этим К. М. Гульдберг и П. Вааге пришли к математическому выражению того эффекта, который в свое время был подмечен Бертолле и назван «действием химических масс», а после работ Гульдберга и Вааге вошел в химию под названием закона действующих масс. Согласно этому закону, как известно, скорость реакции и даже ее направление зависят не только от химической природы веществ, но и от их концентрации, что служит прямым подтверждением идей Бертолле о возможности непрерывного изменения химического сродства в связи с изменением действующих масс. Весьма характерно, что Гульдберг и Вааге открытый ими закон обсуждали на примере реакций, осуществлявшихся в растворах, и при этом неизменно подчеркивали необходимость изучения природы и величины химического сродства. Сами они, исходя из установленной ими зависимости скорости реакций от действующих масс, приходили к выводу, что принципиальной разницы между силами химического сродства в «собственно химических соединениях» и в соединениях переменного состава не существует. «Постепенно открываются новые соединения, которые можно рассматривать как промежуточные члены между этими двумя видами химических соединений, и они доказывают нам, что нельзя провести между ними никакой естественной и строго определенной границы. Рассматривая растворы, теплоты, выделяемые при их образовании, их точки кипения, пришли ко взгляду, что различие между ними чисто в т о р о с т е п е н н о е, а не основное... Мы рассматриваем силы, действующие в этих двух случаях, как силы одного и того же рода».
К таким же выводам пришли В. Харкурт и В. Эссон, впервые в середине 60-х годов осуществившие кинетические исследования окисления йодистого водорода и щавелевой кислоты перманганатом калия в водных растворах.
Таким образом, к 60-м годам XIX столетия был накоплен достаточно большой фактический материал, свидетельствовавший о химической природе взаимодействия между частицами растворителя и растворенного вещества. При этом термин «частицы» в данном случае оказывался, пожалуй, наиболее подходящим, так как было установлено, что взаимодействие в растворе осуществляется не только между молекулами, но и между агрегатами молекул.
Совершенно очевидно, что уже такой вывод не укладывался в рамки химической атомистики Дальтонa, ибо последняя к химическому взаимодействию относила лишь такое, которое происходит между, веществами в строго определенных пропорциях и приводит к соединениям постоянного состава с простыми кратными отношениями. В растворах же, как оказалось, вещества взаимодействуют или в очень широких интервалах пропорций, или в любых пропорциях и образуют соединения пepеменного состава. Химическая атомистика считала, что при образовании соединений действуют прочные химические силы сцепления между атомами и что «сложные атомы», или молекулы, в результате взаимного насыщения в них единиц сродства неспособны взаимодействовать между собою иначе, как только по реакции взаимного обмена. В paстворах же, как было выяснено, действуют слабые химические силы, приводящие к ассоциациям «насыщенных» молекул растворителя и растворенного вещества.
Конечно, исследования первой половины и 60-х годов XIX столетия еще не могли привести к глубокому пониманию сущности растворения. Тогда еще не существовало надежных методов экспериментального изучения растворов, многие исследователи не решались отойти от положений химической атомистики, с которой потребовалось вступать в противоречия; главное внимание в исследованиях поэтому уделялось нахождению в растворах определенных дальтоновских соединений или гидратов.
Дальнейшим развитием химической интерпретации растворов явились исследования Д. И. Менделеева, вылившиеся в фундаментальное строгое учение. Эти исследования были начаты в 60-х годах XIX в. и продолжались до начала ХХ столетия. В 1865 г. Менделеев, «оставляя пока в стороне теоретическую часть, ...обратил... главное внимание на усовершенствование способов исследования и на оценку данных опыта». Он детально изучил зависимость изменения удельного веса спиртовых растворов от состава в интервале температур от 0 до 300 С. Убедившись, как и другие экспериментаторы, в образовании химических соединений в процессе растворения, он встал на позиции синтеза в этом вопросе представлений Бертолле и «великого учения Дальтона об атомном строении вещества». В 1887 г., собрав и обобщив огромный экспериментальный материал, Менделеев опубликовал ставшее широко известным «Исследование водных растворов по удельному весу». Здесь он рассмотрел зависимость изменения удельного веса от изменения состава водных растворов 233 веществ: кислот, щелочей, газообразных веществ, солей и органических соединений.
Построив график такой зависимости для раствора Н2О + SО3, Менделеев указал на существование так называемых «особых точек», или переломов и разрывов сплошных прямых линий, которые могут свидетельствовать о наличии в растворе при определенном процентном содержании SO3 в воде определенного соединения: H2SО4, H2SО4·Н2О, H2SО4·6Н2О и т.д.
Taкого же рода соединения посредством «особых точек» Менделеев обнаружил в водном растворе этилового спирта - С2Н5ОН·Н2О; С2Н5ОН·3Н2О; С2Н5ОН∙12Н2О. В результате этих и других своих работ он пришел к выводу о том, что растворы представляют собою в своей ocнове определенные химические соединения, переменность состава которых обусловлена непрерывной ассоциацией и диссоциацией сольватов, растворенных как друг в друге, так и в избытке растворителя. И хотя этот вывод свидетельствует о том, что Менделеев явно отдавал дань атомистике Дальтона, т. е. хотел подчеркнуть, «что растворы управляются обычными законами химического воздействия, что в них сокрыты те же определенные соединения, которыми так сильна химия, что здесь... существуют свои скачки, свои разрывы сплошности», все же он оставался верен своему тезису, высказанному еще в 60-х годах: «определенные химические соединения составляют только частный случай неопределенных химических соединений». И дело здесь заключается не только в динамизме: и переходном состоянии таких соединений, как H2SО4·Н2О, H2SО4·6Н2О, С2Н5ОН∙12Н2О и т.д., а в том, что сами эти соединения нельзя отнести к определенным дальтоновским соединениям: они непрочны, ибо образуются за счет сил побочного сродства; они противоречат принципу наибольшей простоты, который наряду с другими лег в основу атомистики Дальтона; они не подчиняются законам эквивалентов и простых кратных отношений. Это не гидраты или сольваты, способные существовать в индивидуальном состоянии, как, например, CuSО4∙5Н2О, а молекулярные комплексы, взаимно переходящие друг в друга, находящиеся в условиях непрерывной диссоциации и ассоциации.
Недаром Д. П. Коновалов, ученик Д. И. Менделеева, подчеркивал, что «принимая в основе раствора химизм, нельзя, однако, отождествлять растворение с актом законченного образования определенного соединения» и что «природа раствора определяет... не тем состоянием, в котором вещество выделяется из раствора, а тем, которое оно представляет в растворе».
Принципиальные выводы Д. И. Менделеева о природе растворов были подтверждены в 80-90-х годах ХIХ в. Д. П. Коноваловым, С. Пикерингом, Е. Бироном, Г. Джонсом, А. А. Потылициным и др. Метод, предложенный Менделеевым и заключающийся в изучении растворов по диаграммам состав-свойства, нашел затем широкое распространение. Как подчеркивал Д. П. Турбаба, этот метод был «тем более драгоценным, что позволял открывать вещества, наиболее непрочные из всех химических соединений.
В конце прошлого - начале текущего столетия химическая концепция растворов пополнилась, целым рядом новых положений. Как прямое продолжение ее появилось утверждение об ассоциации молекул и диссоциации комплексов жидкостей:
nH2O↔(H2O)n; nC2H5OH↔(C2H5OH)n
(И. Ван-Лаар, В. Я. Курбатов, В. А. Кистяковский и др.). По существу это утверждение представляло собою вывод из множества экспериментальных работ (С. Пикеринга, Г. Джонса, Д. П. Турбабы, А. В. Сапожникова, А. Г. Дарошевского, Д. П. Ко новалова, Г. Розенбома и других). Согласно этому выводу такие растворы, как спирт + вода, ацетон + вода и т.п., стали рассматриваться как равновесные системы ассоциатов растворителя А, paстворенного вещества В и их комплексов АхВу
Аn + Вm + AxBy↔Аn …. АхВу …. Вm
(n, m, x и у - непрерывно изменяющиеся целые числа) с преимущественным накоплением сольватов определенного состава в особых менделеевских точках.
Совершенно естественным поэтому было появление в конце XIX - начале ХХ столетия ряда гипотез, объясняющих образование в растворах различного рода неопределенны соединений или своеобразных «определенных» молекулярных комплексов типа гидратов, переходящих друг в друга:
MX·nH2O ↔ МХ∙n – mH2O + mH2O.
Не могло уже показаться неожиданным также и то, что в этих гипотезах насыщенные молекулы, даже такие, как Н-О-Н, стали наделяться «добавочным» или «остаточным» сродством.
Ф. М. Флавицкий для объяснения природы растворов и образования гидратных соединений в 1891 г. выдвинул гипотезу, согласно которой некоторые элементы могут проявлять в одних случаях низшую валентность:
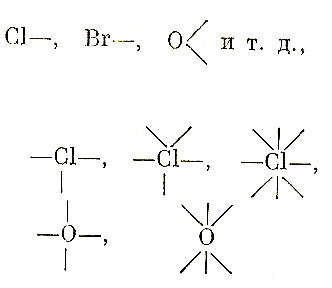
а в других высшую:
При этом «остаточные единицы сродства кислорода или галоидов, например, способны присоединять одну, две и более молекул воды, спирта, кислородсодержащих кислот и т.д. и образовывать гидраты или вообще сольваты неопределенного, т. е. постоянно изменяющегося состава.
С. Пикеринг для объяснения природы растворов выдвинул «теорию der chemischen Residual – Affinität» (химического остаточного сродства). Г. Армстронг для тех же целей предложил близкую к ней гипотезу неполной нейтрализации зарядов при соединении двух разноименно заряженных атомов:
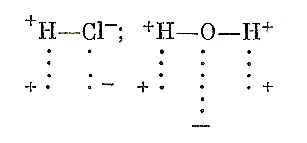
В результате остаточного и к тому же обладающего зарядом сродства насыщенные молекулы растворителя могут направленно (плюс к минусу и наоборот) взаимодействовать между собою:
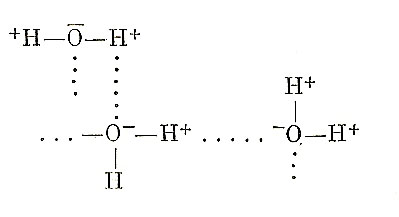
и с молекулами растворенного вещества:
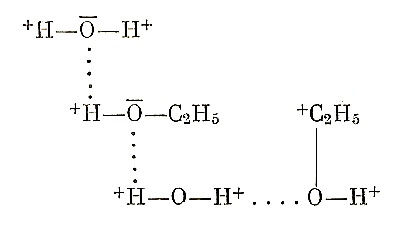
и образовывать комплексы.
Эти и подобные им теории, в особенности такие, как теории Пикеринга и Армстронга, сыграли немаловажную роль в объединении физических теорий растворов с классическими химическими теориями. Легко видеть, как близко стоит только что приведенное объяснение Армстронга к теории ионной гидратации, которая возникла в работах И. А. Каблукова, В. А. Кистяковского, Л. В. Писаржевского, А. Вернера, Р. Абегга, Ф. Кольрауша и других исследователей тоже в 90-х годах прошлого столетия. Эта теория объединила представления Аррениуса об электролитической диссоциации, казалось бы, с противоположной этому химической концепцией Менделеева, Пикеринга, Джонса и других. Согласно теории ионной гидратации, как известно, диссоциация в водных растворах вызывается первоначальным образованием гидратов электролитов
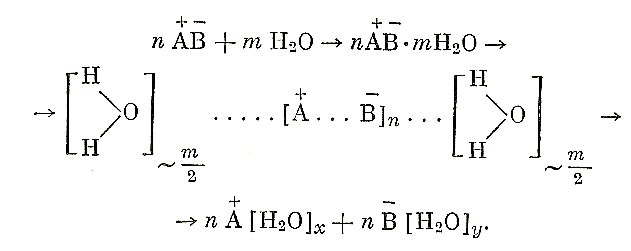
Образующиеся в результате этого ионы остаются также гидратированными. При этом величины n, m, х и у являются, естественно, целочисленными, но далеко не постоянными; они зависят прежде всего от энергии связи А-В как в кристаллической решетке, так и в молекулярном состоянии, а кроме того, от ионных радиусов А и В и от условий растворения.
Конечно, еще не все проблемы теории растворов решены и в настоящее время. Но многочисленные физические методы, широко используемые теперь в самых различных областях химии, и применительно к растворам позволили прийти к выводу, что представления, развиваемые в недрах классической химии, не были лишены основания. Высказанные ранее предположения, базирующиеся больше на интуиции, чем на открытом логическом обобщении экспериментальных данных, в основном оказались достоверными. Растворы, действительно, как это установлено, являются неопределенными химическими соединениями, образованными за счет тех относительно слабых химических связей, которые в общей форме можно назвать неполновалентными. Если эти связи гомеополярные, то они обладают электронным зарядом (или «электронной заселенностью») менее одного электрона, а если они ионные или диполь-дипольные, то их энергия составляет обычно менее половины той, которая присуща ионной связи при наличии почти полного перехода электрона от атома к атому (например, Na + Cl → Na+ - Cl-).
Современная теория растворов, построенная на основе богатейшего собственно химического материала, а также данных термохимии, электрохимии, рентгенографии, спектроскопии, дифракции нейтронов, изотопного обмена и других физических и физико-химических методов исследования, предусматривает значительно больший комплекс факторов, влияющих на растворение и на свойства растворов, чем это имело место в классической химии. К таким факторам относятся: химическое строение молекул растворителя и растворимого вещества, структура и энергетическое состояние того и другого вещества в конденсированном состоянии (для растворителя - в жидком состоянии, а для растворимого вещества в жидком, твердом и иногда в газовом), термодинамические условия, влияние примесей и стенок сосуда и т.д.
Дата добавления: 2016-05-30; просмотров: 1796;
Поиск по сайту
Узнать еще
- IV. Права и обязанности личного состава службы
- V. Подготовка личного состава службы
- VI. Тормоза подвижного состава
- VIII. Сигналы, применяемые для обозначения поездов, локомотивов и другого железнодорожного подвижного состава
- Амфотерность соединений
- Анализ гармонического состава выходного напряжения.
- Анализ гармонического состава потребляемого тока.
- Анализ динамики, состава и структуры источников формирования капитала предприятия

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
