Нарушение транспорта липидов
Всосавшиеся в кровь неполярные липидные молекулы циркулируют в крови и лимфе в комплексе с полярными соединениями
(белками). Существует большой спектр липопротеиновых частиц, отличающихся по размерам, плотности и составу. Липопротеины- это сферические частицы, состоящие из гидрофобной сердцевины и гидрофильной оболочки (в центре - неполярные липиды: триглицериды и эфиры холестерина; оболочка построена из полярных липидов - холестерина и фосфолипидов, причем их заряженные концы обращены наружу; также в составе оболочки - белки, нековалентно связанные с фосфолипидами и холестерином, апопротеины). Апопротеиныподдерживают структуру частиц, обеспечивают их взаимодействие с рецепторами и служат «визитной карточкой» липопротеинов, поскольку рецепторы для липопротеинов на разных клетках распознают только определенные апопротеины. Липопротеины подразделяют на классы в зависимости от их плотности и подвижности при электрофорезе. Плотность липопротеиновой частицы определяется отношением апопротеины/ липиды: чем больше это отношение, тем выше плотность. Выделяют 4 основные группы липопротеинов (ЛП):
1. Липопротеины высокой плотности (ЛПВП, илиα-ЛП). В состав ЛПВП входят 40-55% белка (процент общей массы частицы), 27-30% фосфолипидов, 3-8% триглицеридов, 2-3% свободного холестерина, 14-20% эфиров холестерина. Оболочка содержит апопротеины А, СП, Е.Они синтезируются паренхимой печени, в стенке тонкого кишечника и всегда присутствуют в плазме крови здоровых людей. Выполняют транспортную функцию, переводя избыток холестерина с поверхности сосудов в печень и выводя его излишек из клеток эндотелия (рис. 12-29).
Высокоспецифичные рецепторы ЛПВП обнаружены на гладкомышечных клетках и фибробластах. Количество этих рецепторов увеличивается при повышении концентрации холестерина в клетке. Связывание ЛПВП с рецепторами вызывает выброс холестерина из клеток. Первоначально холестерин встраивается в оболочку ЛПВП, но затем под действием лецитин-холестеринацетилтрансферазы (ЛХАТ) он этерифицируется и перемещается в сердцевину ЛПВП. В печени ЛПВП связываются с рецепторами и разрушаются. Таким образом, ЛПВП - это антиатерогенные липопротеины.
2. Липопротеины очень низкой плотности (ЛПОНП, или пре-β-ЛП).Представляют очень неоднородный класс частиц с различным содержанием компонентов: 8-12% - белок, 10-12% - свободный холестерин, 18-20% - фосфолипиды, 3-6% - эфиры
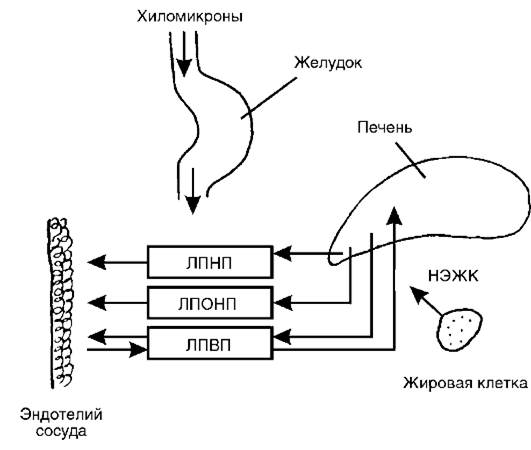 Рис. 12-29.Обмен липидов. ЛПНП - липопротеины низкой плотности, ЛПОНП - липопротеины очень низкой плотности, ЛПВП - липопротеины высокой плотности, НЭЖК - неэтерифицированные жирные кислоты
Рис. 12-29.Обмен липидов. ЛПНП - липопротеины низкой плотности, ЛПОНП - липопротеины очень низкой плотности, ЛПВП - липопротеины высокой плотности, НЭЖК - неэтерифицированные жирные кислоты
холестерина, около 50% - триацилглицеролы (ТАГ). Они образуются в основном в гепатоцитах (и в меньшем количестве - в слизистой кишечника), являются главной транспортной формой эндогенных триацилглицеролов. В их составе апопротеины С, Е и В100.В плазме крови происходит трансформация ЛПОНП в β-ЛП (при участии ферментов - липопротеиновой липазы и ЛХАТ крови). В ходе их катаболизма размеры частиц уменьшаются, меняется их состав (теряются ТАГ и возрастает относительный процент холестерина).
3. Липопротеины низкой плотности (ЛПНП, илиβ-ЛП)имеют следующий состав: 24-31% - свободный холестерин, 16-28% - этерифицированный холестерин, 7-11% - триглицериды, около 30% - фосфолипиды, 20-25% - белок. Они образуются в плазме из ЛПОНП и являются самой атерогенной фракцией липопротеинов у человека. ЛПНП содержат только один апопротеин B100.
Около 70% ЛПНП удаляется из крови гепатоцитами с помощью высокоспецифичных рецепторов. Остальные ЛПНП захватываются клетками ретикулоэндотелиальной системы с помощью менее
специфичных нейтрализующих рецепторов. Синтез высокоспецифичных рецепторов в печени подавляется при повышении концентрации β-ЛП. Напротив, количество нейтрализующих рецепторов на клетках ретикулоэндотелиальной системы не зависит от уровня ЛПНП. Нарушение захвата ЛПНП в печени (вследствие дефекта апопротеина B100 или дефекта высокоспецифичньгх рецепторов) приводит к накоплению ЛПНП в других тканях и органах. Их атерогенный эффект опосредуется именно нейтрализующими рецепторами. Считается, что ЛПНП становятся атерогенными только после определенных превращений, например, при перекисном окислении (модифицированные ЛПНП). Окисленные ЛПНП захватываются макрофагами, которые после этого превращаются в ксантомные клетки, нагруженные эфирами холестерина. Нейтрализующие рецепторы обнаружены и в гладкомышечных клетках артерий.
4. Хиломикроны- самые крупные липопротеиновые частицы, поступающие в кровь из лимфы и представляющие собой транспортную форму пищевых жиров (экзогенных ТАГ). В их составе находятся: 3-8% фосфолипидов, 2-4% эфиров холестерина, около 2% свободного холестерина, 1-2% белка и 86-94% ТАГ. В оболочке имеются апопротеины B48, A, C, E.Хиломикроны образуются в стенке кишечника в процессе всасывания экзогенных ТАГ и холестерина, поступают в лимфатические сосуды и через грудной лимфатический проток попадают в большой круг кровообращения.
В крови хиломикроны встречаются с ЛПВП и обмениваются с ними апопротеинами: отдают часть апопротеинов A, получают C и E. В кровеносных капиллярах жировой ткани, миокарда, скелетных мышц и молочных желез хиломикроны расщепляются липопротеиновой липазой, расположенной на поверхности эндотелия и теряют значительное количество ТАГ (образуются свободные жирные кислоты (СЖК) и глицерин). При этом освобождается остаточный компонент хиломикрона (сердцевина), содержащий большое количество эфиров холестерина. Сами хиломикроны не обладают атерогенными свойствами, но остаточные компоненты хиломикронов, по-видимому, атерогенны.
Для ткани легких катаболизм хиломикронов особенно важен, поскольку играет ключевую роль в обеспечении высокой активности альвеолярных макрофагов и необходим для синтеза фосфолипидов сурфактанта (рис. 12-30). В связи с этим при заболеваниях легких положительный эффект дает жировая диета. Следует от-
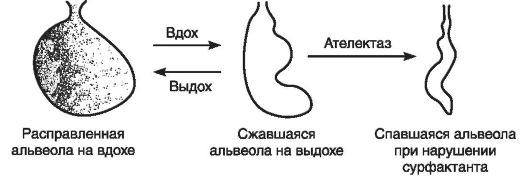 Рис. 12-30.Роль сурфактанта в предупреждении ателектаза
Рис. 12-30.Роль сурфактанта в предупреждении ателектаза
метить, что плазма крови здоровых людей натощак (через 12-14 ч после приема пищи) не содержит хиломикронов. В крови, взятой натощак, присутствуют только ЛПОНП, ЛПНП и ЛПВП, тогда как другие липопротеины (хиломикроны, остаточные компоненты хиломикронов, а также липопротеины промежуточной плотности) выявляются только после еды или при нарушениях обмена липидов. При ряде заболеваний липопротеиновый спектр сыворотки меняется и возникают гиперили гипо-, алипопротеинемии. При этом отмечаются увеличение или, наоборот, снижение содержания, вплоть до полного отсутствия одного или нескольких классов липопротеинов в крови, а также появление их определенных форм (дислипопротеинемии). Гипер- и дислипопротеинемии - один из главных факторов риска многих заболеваний, в первую очередь - атеросклероза. Их клинические проявления и тяжесть зависят от рациона и режима питания, а также от сопутствующих заболеваний. Дислипопротеинемии возникают или усиливаются при ожирении, сахарном диабете, гипотиреозе, болезнях почек и печени; их течение и прогноз зависят от тяжести основного заболевания. Различают 5 типов гиперлипопротеинемий:
I. Гиперхиломикронемия- характеризуется высоким содержанием хиломикронов в плазме натощак. Проявляется ксантоматозом- отложением холестерина и его эфиров в купферовских клетках печени, гистиоцитах подкожной клетчатки и сухожилиях с последующим разрастанием соединительной ткани в виде бляшек и узлов желтоватого цвета (рис. 12-31). У больных развивается гепатоспленомегалия, выявляются тромбоз и микронекрозы поджелудочной железы с последующим формированием хронического панкреатита, абдоминальные колики после принятия жирной пищи. На коже определяются ксантомы в виде желтоватых папул. Заболе-
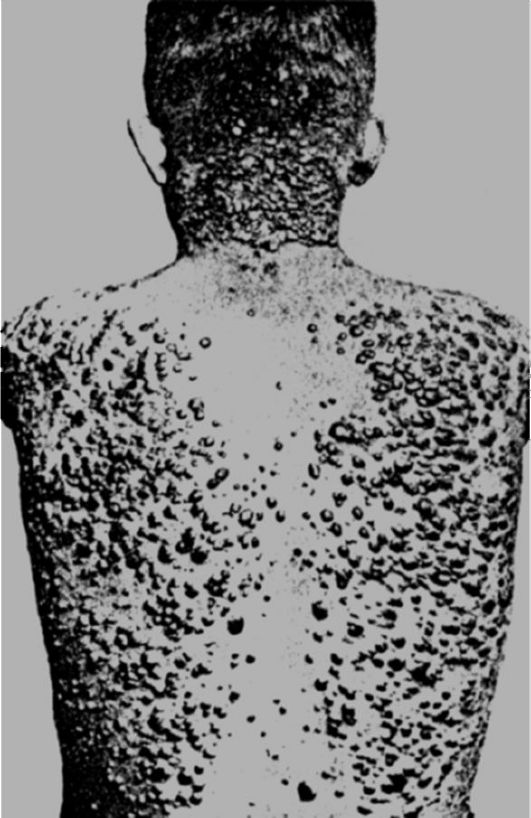 Рис. 12-31.Ксантоматоз кожи (по H.W. Siemens)
Рис. 12-31.Ксантоматоз кожи (по H.W. Siemens)
вание может быть вызвано наследственным аутосомно-рецессивным дефектом липопротеиновой липазы либо аутоиммунными заболеваниями соединительной ткани (при системной красной волчанке образуются антитела против гликозаминогликанов, что нарушает процесс гепариновой активации липопротеиновой липазы).
II. Гипер-b-липопротеинемия делится на 2 типа:
11а - увеличение содержания в крови β-ЛП при нормальном уровне пре- β-ЛП;
11б - увеличение содержания β-ЛП и пре- β-ЛП.
Для заболевания характерен выраженный ксантоматоз век, кожи, роговицы, развитие ишемической болезни сердца с инфарктом миокарда в очень раннем возрасте,
атеросклеротические поражения сосудов у детей. Предполагается, что в основе заболевания лежит аутосомно-доминантный дефект рецепторов ЛПОНП и ЛПНП (11б) либо изменение активности липопротеиновой липазы плазмы крови (11а).
III. «Флотирующая» гиперлипопротеинемия, или дис- β-липопротеинемия.В основе заболевания лежит наследственно обусловленное нарушение синтеза апопротеина Е (белок, входящий в состав хиломикронов и ЛПОНП). Заболевание характеризуется появлением в сыворотке флотирующихβ-ЛП, которые называются промежуточными (липопротеины промежуточной плотности- ЛППП).Они обогащены холестерином, а содержание триглицеридов в них может быть снижено. Образуются эти частицы при нарушении катаболизма ЛПОНП и хиломикронов. Встречаются также приобретенные формы заболевания при гипотиреозе, танжерской болезни, некоторых аутоиммунных гаммапатиях. Этот вид гиперлипопротеинемии сопровождается ранними атеросклеротическими проявлениями (после 20 лет), развитием ишемической болезни сердца, ишемической энцефалопатии вплоть до инсультов, ксантоматозом, ожирением.
IV. Гипер-пре- β-липопротеинемия.Заболевание может быть наследственно обусловленным (аутосомно-доминантное) или приобретенным (при алкоголизме, остром гепатите, акромегалии, диабете и др.). Патогенез до конца не выяснен. Для этого типа гиперлипопротеинемии характерно нарастание уровня триглицеридов и ЛПОНП в крови. Содержание ЛПНП и ЛПВП варьирует от нормального до значительно сниженного. У больных развиваются ожирение и сахарный диабет 2 типа, появляются ксантомы, возможны атеросклеротическое поражение сосудов нижних конечностей, липидоз сетчатки и ухудшение зрения, проявления ишемической болезни сердца.
V. Гипер-пре- β-липопротеинемия и хиломикронемия.При этом заболевании в крови увеличивается содержание хиломикронов и ЛПОНП и снижается уровень ЛПНП и ЛПВП. У больных отмечаются гепато- и спленомегалия, ожирение, снижение толерантности к глюкозе (при сахарном диабете 2 типа), поражение миокарда. После приема жирной пищи могут наблюдаться внезапные приступы абдоминальной колики, ксантоматоз и атеросклероз слабо выражены. В патогенезе первичного заболевания главную роль играет наследственно обусловленное отсутствие кофактора липопротеинлипазы - апопротеина CII (аутосомно-рецессивное наследование), в результате два основных субстрата воздействия этого фермента накапливаются в крови. Фенокопия болезни развивается при алкоголизме, гликогенозе Гирке и некоторых других заболеваниях печени.
Гипо-(а)-липопротеинемии относятся к группе относительно редких аномалий спектра липопротеинов:
1. A-β-липопротеинемия.В основе заболевания лежит аутосомно-доминантный дефект синтеза апопротеина В, что приводит к аномалии строения хиломикронов, снижению содержания или полному отсутствию в плазме ЛПОНП и ЛПНП. Клинические проявления связаны с нарушением всасывания в кишечнике жиров и углеводов, гемолитической анемией, дегенерацией бокового и заднего канатиков спинного мозга, пигментной ретинопатией. Нарушение всасывания жиров проявляется сразу после рождения плохим аппетитом, рвотой, обильными испражнениями, стеатореей, развитием гипотрофии. Примерно у трети больных развивается умственная отсталость. С возрастом усиливаются неврологические расстройства, появляются скелетные деформации, сердечные аритмии, ухудшается зрение. В патогенезе заболевания решающее значение имеет снижение содержания холестерина в клеточных
мембранах и потеря жирорастворимых витаминов, особенно витамина Е, что ведет к утрате антиоксидантной защиты мембран.
2. Танжерская (или тэнжирская) болезнь.В основе заболевания лежит аутосомно-рецессивное нарушение синтеза апопротеина А, что, в свою очередь, нарушает продукцию ЛПВП. У больных нарушен транспорт эфиров холестерина, в результате эфиры захватываются макрофагами и откладываются в клетках ретикулоэндотелиальной системы селезенки, печени, лимфоидных органов. Выявляются лимфаденопатия, гепатоспленомегалия, неврологические нарушения - слабость, парестезии, снижение сухожильных рефлексов. Одним из ярких признаков заболевания является оранжево-желтый цвет увеличенных миндалин.
Существуют и другие формы гиполипопротеинемий: церебросухожильный ксантоматоз (наследственный дефект синтеза желчных кислот из холестерина), болезнь Вальмана (аутосомнорецессивный дефицит холинэстеразы), гипо-a-липопротеинемия (генетически детерминированное нарушение продукции апопротеина А и С) и др. Большинство из них связано с наследственной патологией синтеза белковой части липопротеинов либо с нарушением метаболизма холестерина.
Дата добавления: 2016-07-11; просмотров: 3830;
Поиск по сайту
Узнать еще
- V Патопсихологическое – при нарушении целостности мозга происходит нарушение психической деятельности
- Агрегативная устойчивость растворов ВМС и ее нарушение
- Биологическая роль липидов.
- БИОЛОГИЧЕСКИЕ ФУНКЦИИ ЛИПИДОВ
- Биосинтез липидов и их компонентов.
- Биосинтез полярных и неполярных липидов
- Биосинтез фосфолипидов
- Биофизические механизмы транспорта вещества через биомембраны.

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
