Реакции, катализируемые полупроводниковыми оксидами
Представления о хемосорбции на поверхности полупроводниковых оксидах будут полезны для понимания некоторых закономерностей катализа в реакциях окисления и дегидрирования.
Реакции окисления на полупроводниковых оксидах состоят из нескольких стадий:
1. Хемосорбция субстрата (т.е. образование связей окисляемого вещества с катализатором);
2. Хемосорбция кислорода;
3. Передача электронов от окисляемого субстрата (донора) к кислороду (акцептору) через катализатор;
4. Взаимодействие образующихся из субстрата ионов, радикалов, ион-радикалов на поверхности катализатора с анионами кислорода, приводящее к образованию промежуточных или конечных продуктов реакции;
5. Возможная перегруппировка промежуточного продуктов;
6. Десорбция продуктов окисления.
Как было показано выше (зонная схема), оксиды р-типа способны адсорбировать кислород в гораздо большем количестве, чем оксиды n-типа. Это объясняет тот факт, что оксиды р-типа более активные катализаторы окисления. Однако очень часто активность и селективность меняются в противоположных направлениях, и бóльшая адсорбция кислорода на оксидах р-типа (т.е. большая активность в реакциях окисления) приводит к полному окислению субстрата (например углеводородов до Н2О и СО2) т.е. к низкой селективности. Применение же оксидов n-типа позволяет проводить селективное окисление.
Соотношение адсорбированных молекул (кислород)/(углеводород) на оксидах р-типа очень высоко и практически не регулируется изменением парциального давления О2.
Напротив, в случае оксидов n-типа концентрация активированного кислорода на поверхности может регулироваться парциальным давлением, а еще эффективнее природой и количеством модифицирующих добавок в оксиде, что позволяет проводить селективное окисление.
Различия в селективности окисления объясняют следующими факторами:
- Различием в электроотрицательности металлов оксида;
- Потенциалом ионизации металлов оксида;
- Силой связывания кислорода поверхностью катализатора (способностью к отщеплению кислорода решетки).
Для описания многих промышленных реакций окисления чаще всего используют два простых двухстадийных механизма, представленных на Рисунке 5.48 схемами А и В.
А: 0,5О2(Г) à О*
Х* + О* à продукты
В: Х* + О(РЕШЕТКА) à продукты + вакансия в решетке
0,5О2(Г) + вакансия в решетке à О(РЕШЕТКА)
Рис. 5.48. Механизмы окисления. Х - окисляемый субстрат.
По механизму А кислород гораздо быстрее и в больших концентрациях чем субстрат (Х) адсорбируется на поверхности. Адсорбирующийся субстрат (Х*) реагирует с избыточным кислородом (О*) на поверхности. Такой механизм предполагает полное окисление субстрата до Н2О и СО2.
По механизму В (механизм Марса-Ван-Кривелена) молекула адсорбированного субстрата (Х*) реагирует с кислородом решетки катализатора. Такой механизм приводит к селективному (частичному) окислению (например, окисление пропилена до акролеина).
Селективные катализаторы окисления, работающие по механизму Марса-Ван-Кривелена, как правило являются оксидами n-типа, в которых катионы металла имеют полностью пустые или полностью заполненные d-орбитали:
Mo6+ V5+ Sb5+ Sn4+ Bi3+
4d0 3d0 4d10 4d10 5d10
Металлы, находящиеся в высшей степени окисления (оксиды n-типа) легко теряют кислород решетки, формально в виде О-2, восстанавливаясь до низших степеней окисления. Для такого хорошо известного катализатора селективного окисления, как V2O5, установлено, что скорость окисления на нем H2,CO ,CH2=CH2, бутена, бутадиена и ксилола зависит от концентраций связей V=O на поверхности катализатора.
Решающей стадией является восстановление V5+ до V4+ с переходом кислорода из решетки к окисляемому реагенту:
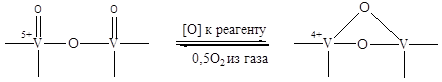 (5.49)
(5.49)
По подобному механизму протекает окисление метанола до формальдегида на центрах Мо=О оксида молибдеда.
Если посмотреть на приводимые в научной литературе ряды относительной селективности оксидных катализаторов в реакциях окисления:
окисление пропилена до акролеина
MoO3 > Sb2O5 > V2O5 > TiO2 = Fe2O3 > SiO2
окисление бензола до малеинового ангидрида
V2O5 > Cr2O3 > MoO3 > Co2O3
окислительная циклодимеризация пропилена до 1,3-гексадиена
ZnO > Bi2O3 > In2O3 > SnO2 > Ga2O3 > CdO
то становится очевидно, что не существует единого ряда селективности катализаторов для всех реакций окисления.
Связь электронного строения полупроводника с его каталитическими свойствами удобнее всего наблюдать на примере относительно простых, модельных, реакций. Рассмотрим несколько таких примеров.
Разложение N2O.
Реакция разложения N2O протекает по следующему уравнению:
2N2O à 2N2 + O2 (5.50)
Данная реакция катализируется множеством оксидов. Взяв за меру каталитической активности температуру начала разложения, исследованные оксиды можно расположить в следующий ряд по активности:
| Т, оС | 200 – 300 | 300 – 600 | 600 - 750 |
| Cu2O, CoO, NiO | CuO, MgO, CaO, CeO, Al2O3 | ZnO, CdO, TiO2, Cr2O3, Fe2O3, Ga2O3, | |
| Полупроводники р-типа | Изоляторы | Полупроводники n-типа |
Механизм протекания данной реакции следующий:
N2O + е à N2 + O-(адс.) (5.51)
O-(адс.)+ N2O à N2 + O2 + e
На первой стадии происходит передача электронов от катализатора к атомам кислорода. Как было показано выше, оксиды р-типа способны к хемосорбции кислорода в гораздо большей степени, чем оксиды n-типа. Лимитирующей в данном процессе является вторая стадия – передача электрона катализатору и десорбция продуктов. O-(адс.) можно рассматривать как кислород решетки, поэтому его электрон располагается в валентной зоне (см. Рис. 5.41 и 5.42). Чтобы быть переданным одному из атомов катализатора, электрон должен преодолеть энергетический барьер для перехода на локальный акцепторный уровень (в случае р-полупроводника) или в зону проводимости (в случае n-полупроводника). То есть, активность катализатора определяется уровнем Ферми (который должен быть в данном случае меньше энергии ионизации O-(адс.)). В соответствии с зонной схемой полупроводников, энергетический барьер для передачи электрона катализатору от O-(адс.) в случае оксидов р-типа ниже, чем в случае оксидов n-типа. Поэтому большую активность в данной реакции проявляют оксиды р-типа, а наименее активны – оксиды n-типа.
Увеличение дырочных дефектов в оксиде р-типа также должно приводить к увеличению скорости этой реакции. Так и происходит при добавлении в NiO примеси Li2O в количествах до 0,1 %. При этой концентрации модификатора наблюдается максимум активности, а далее активность падает. Это объясняется тем, что при большем количестве дырок уже сильно затруднена адсорбция N2O, то есть первая стадия процесса. Добавка Cr2O3 к NiO, напротив, приводит к снижению дырочных дефектов и, соответственно, к снижению скорости данной реакции.
Окисление СО.
Еще одной подробно изученной модельной реакцией является окисление СО. Результаты исследований каталитической активности модифицированных и немодифицированных NiO и ZnO приведены в Таблице 5.18.
Таблица 5.18.
Окисление СО на оксидных полупроводниковых катализаторах
| Катализатор | Энергия активация, кДж/моль |
| NiO (р-тип) + Cr2O3 + Li2O | |
| ZnO (n-тип) + Cr2O3 + Li2O |
Сравнение величин энергий активации показывает, что полупроводник р-типа (NiO) активнее, чем полупроводник n-типа (ZnO). С точки зрения зонной теории это означает, что лимитирующей стадией реакции является адсорбция донорной молекулы СО с передачей электрона от нее к катализатору (в полупроводнике р-типа передача электрона дырке).
CO + [+] à CO+(адс.) (5.52)
Соответственно, чем больше дырочных дефектов у NiO, тем выше скорость данной реакции. Поэтому добавка Li2O (повышающая дырочную проводимость) ускоряет реакцию, а добавка Cr2O3 (понижающая дырочную проводимость) – замедляет (Табл. 5.18). Кинетические исследования подтверждают вывод о лимитирующей стадии (5.52), так как наблюдается первый порядок по СО.
Адсорбция кислорода (акцептора) на NiO проходит быстро (так как Ni2+ лекго окисляется до Ni3+) и скорость реакции не зависит от давления кислорода:
0,5 O2 à O-(адс.) + [+] (5.53)
Завершающая стадия реакции протекает по механизму Лэнгмюра-Хиншельвуда:
CO+(адс.) + O-(адс.) à CO2 (газ) (5.54)
Кинетические эксперименты показали, что на полупроводнике n-типа (ZnO) скорость реакции сильно зависит от давления кислорода. Это означает, что акцепторная хемосорбция кислорода (5.53) так же лимитирует скорость процесса. Это объясняет влияние модифицирующих добавок на активность ZnO. Увеличение количества донорных дефектов (добавлением Cr2O3) приводит к увеличению активности, а снижение количества донорных дефектов (добавлением li2O) – к снижению.
Приведенный пример показывает, что при переходе от одного типа полупроводника к другому может изменяться механизм реакции.
Каталитическое разложение этилового спирта.
Рассмотрим селективность каталитического превращения этанола, которое может протекать, в зависимости от используемого катализатора, по двум направлениям - дегидрирование и дегидратация:
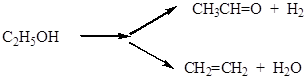 (5.55)
(5.55)
В Таблице 5.19 суммированы результаты экспериментов по изучению влияния природы оксидного катализатора на соотношение образующихся продуктов.
Таблица 5.19.
Разложение этанола на оксидных катализаторах
| Катализатор | Ацетальдегид (%) | Этилен (%) | ||
| Al2O3 Cr2O3 TiO2 ZrO2 Fe2O3 ZnO | 
| Увеличение n-характера оксида | 1,5 | 98,1 |
Данные таблицы показывают, что доля реакции дегидрирования увеличивается с ростом n-характера оксидов и повышением уровня Ферми. Это объясняется тем, что реакция дегидрирования протекает через разрыв связи О-Н с образованием поверхностного аниона C2H5O- за счет акцептирования электрона с донорных уровней катализатора. Поэтому, по мере увеличение n-характера оксидов растет доля реакции дегидрирования. Для реакции дегидратации решающей стадией является электроно-донорная хемосорбция кислорода спиртовой группы за счет свободной пары электронов, поэтому с ростом р-характера оксидов увеличивается доля реакции дегидратации.
Приведенные примеры показывают, что применение простой зонной модели бывает полезно для объяснения влияния электронного строения полупроводниковых оксидов на особенности катализа в тех или иных реакциях.
Помимо электронного фактора на активность катализатора влияет и много других факторов, в том числе геометрический и энергетический. Поэтому разработка эффективных катализаторов является во многом эмпирической задачей, но с опорой на известные теоретические представления.
На практике наиболее эффективными полупроводниковыми катализаторами являются смешанные оксиды. Наиболее важные промышленные процессы, катализируемые полупроводниковыми оксидами, приведены в Таблице 5.20.
Таблица 5.20.
Промышленные процессы, катализируемые смешанными оксидами
| Процесс | Оксиды металлов |
| Окисление метанола до формальдегида Селективное гидрирование Гидродесульфурирование, гидроденитрование Синтез метанола Окисление бензола до малеинового ангидрида Окислительный аммонолиз пропилена | Fe/Mo, Fe/W Cu/Cr Co/Mo, Ni/Mo Zn/Cr, Zn/Cu V/Mo Bi/Mo, U/Sb |
Так, например, эффективным катализатором синтеза метанола является смешанный оксидный катализатор Zn/Cu. Установлено, что ионы Cu1+, находящиеся в решетке ZnО хемосорбируют молекулы СО. Поверхностные ZnО-центры диссоциативно хемосорбируют водород. Гетеролитически распавшийся водород реагирует с хемосорбированным СО с образованием фрагмента -СН-ОН, который далее гидрируется до метанола.
Основываясь на большом практическом опыте, можно сформулировать некоторые эмпирические правила для катализа полупроводниковыми оксидами:
1. Оксиды переходных металлов эффективно катализируют реакции окисления и дегидрирования.
2. Оксиды, в которых металл имеет несколько устойчивых степеней окисления, наиболее активны.
3. Щелочи обычно стабилизируют высшие степени окисления металла в оксиде, а кислоты – низшие.
4. В реакциях каталитического окисления изменения активности и селективности обычно противоположны.
5. Селективные оксидные катализаторы окисления как правило содержат ионы металлов с электронной структурой d0 и d10.
6. Активность катализатора обычно коррелирует с:
- силой связывания кислорода поверхностью;
- DH образования оксида металла;
- доступностью кислорода решетки.
7. Каталитическая активность в реакции окисления Н2, СО и углеводородов коррелирует с энергией связывания поверхностного кислорода:
Co3O4>MnO2>NiO>CuO>Cr2O3>Fe2O3>ZnO>V2O5>TiO2>Sc2O3
Дата добавления: 2020-07-18; просмотров: 537;
Поиск по сайту
Узнать еще
- I тип реакций. Реакции, характерные для органических кислот.
- II период – 1812-1825 гг. – период реакции, получивший название «аракчеевщина».
- Кислоты также вступают в реакцию обмена с оксидами металлов.
- Мономолекулярные реакции, в которые могут вступить радикалы
- Окисление пероксидом водорода и гидропероксидами
- Определение термодинамических параметров реакции, протекающей в гальваническом элементе
- Реакции в ПРПП, катализируемые ферментами

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
