Комплексные соединения
Состав, строение и свойства соединений, которые позднее назвали комплексными (или координационными), в свое время не вписывались в сложившиеся представления химической теории. Так, некоторые вещества, способные к самостоятельному существованию, оказались способными соединяться друг с другом, хотя валентные возможности атомов, казалось, уже исчерпаны. Например, FeF3 и KF способны образовать соединение состава FeF3*3KF, а CuSO4 и NH3 - соединение CuSO4*4NH3. Причем, если ионы Cu2+ и Fe3+ в растворах CuSO4 и FeF3, соответственно, легко обнаружить с помощью реакций обмена (например, действием щелочи), то в приведенных выше их «комплексах» наличие Cu2+ и Fe3 тем же способом не обнаруживается.
Были непонятными, с позиций обычных обменных реакций, некоторые ионно-молекулярные взаимодействия. Так, при добавлении концентрированной соляной кислоты к голубому раствору CuSO4 окраска его менялась на зеленую. Розовый раствор CoCl2 при его выпаривании становился синим без добавления каких-либо реактивов. – Какие «слабые электролиты» при этом образовались? Более того, оказались возможными реакции, прямо противоположные «привычным» обменным взаимодействиям. Например, при смешивании Cu(OH)2 и аммиака, осадок не образовывался, а растворялся, и вместо двух слабых оснований получалась щелочь.
И, наконец, были получены вещества одинакового состава, но разного строения – изомеры. Ранее свойство изомерии было известно только для органических соединений.
Систематизировать и осмыслить перечисленные выше свойства и явления в течение более чем 100 лет пытались многие ученые, но наиболее полно это удалось сделать швейцарскому химику А.Вернеру (за что впоследствии ему была присуждена Нобелевская премия).
Основные положения координационной теории
Согласно теории А.Вернера отличительной особенностью координационных соединений является наличие центрального атома(иона) металла, который называют комплексообразователем. Типичные комплексообразователи – катионы металлов побочных подгрупп: Ag+, Cu2+, Fe3+ и др.
В ближайшем окружении центрального атома располагаются ковалентно с ним связанные анионы кислот или электронейтральные молекулы, которые называют лигандами. Типичные лиганды – анионы кислот (галогенид-ионы, CN-, NO2-), молекулы NH3, H2O и др.
Число связей, образуемых центральным атомом с лигандами, называют его координационным числом (КЧ), а число связей, образуемых каждым лигандом с центральным атомом называют дентатностьюлиганда.
Примеры монодентатных лигандов: Cl–, F–, Br–, I–, CN–, OH–, NH3, H2O.
К числу бидентатных лигандов относится H2N–CH2–CH2–NH2 – этилендиамин, оксалат-, карбонат-ионы и др.
Примером полидентатных лигандов может служить этилендиаминтетраацетат-ион (ЭДТА).
Примечание: КЧ центрального атома – сложная функция, зависящая от размера и заряда центрального атома и лигандов, но оно обычно в 2 раза больше заряда комплексообразователя; уточнить его, также как и наиболее характерные для него лиганды можно с помощью справочника (см. далее – «константы нестойкости»).
Центральный атом с координированными вокруг него лигандами образуют внутреннюю сферукомплекса. При написании полной формулы комплекса внутреннюю сферу выделяют квадратными скобками.
Заряд внутренней сферы равен алгебраической сумме зарядов центрального атома и всех лигандов. Например: [FeF6]3-, [Ag(NH3)2]+, [Co(H2O)4Cl2]
Если заряд внутренней сферы не равен нулю, то имеется внешняя сфера, состоящая из ионов с противоположным знаком (эти ионы могут быть также комплексными). Последовательность записи формулы комплекса определяется зарядами внешней и внутренней сфер: сначала пишут катион, затем – анион. Например: K3[FeF6], [Ag(NH3)2]2(SO4), [Co(NH3)6][Cr(CN)6]. Внутренняя сфера комплекса может быть смешанной (включать не одинаковые, а разные лиганды): [Co(NH3)5Cl]SO4.
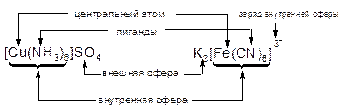
Изомерия комплексных соединений
Изомерия проявляется в существовании соединений с одинаковым составом, но различным строением, вследствие чего эти соединения обладают разными свойствами.
Геометрические (или цис-, транс-) изомеры
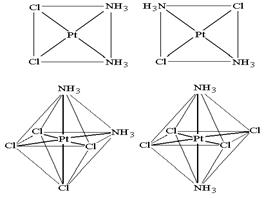
Цис- или транс- изомерия возможна для комплексов со смешанной внутренней сферой квадратного или октаэдрического строения и обусловлена различным пространственным расположением пары одинаковых лигандов относительно центрального атома. Если одинаковые лиганды располагаются в соседних вершинах квадрата или октаэдра, то такой изомер называется цис-изомером, если в противоположных – транс-изомером (рис. 9.1).
|
Гидратные изомеры различаются распределением молекул воды между внутренней и внешней сферами:
[Cr(H2O)6]Cl3 « [Cr(H2O)5Cl]Cl2×H2O « [Cr(H2O)4Cl2]Cl*2H2O
Ионизационные изомеры различаются распределением лигандов - анионов между внутренней и внешней сферами:
[Co(NH3)4Br2]Cl и [Co(NH3)4BrCl]Br; [Pt(NH3)4Cl2]Br2 и [Pt(NH3)4Br2]Cl2
Координационная изомерия возможна для комплексных соединений, у которых и катион и анион комплексные, и обусловлена переходом лигандов от одного комплексообразователя к другому: [Co(NH3)6][Cr(CN)6] « [Cr(NH3)6][Co(CN)6] .
Классификация комплексов
а) классификация по заряду внутренней сферы
Как отмечалось ранее, заряд внутренней сферы комплекса равен алгебраической сумме зарядов комплексообразователя и лигандов. Если этот заряд положительный, то комплекс относят к катионным, если отрицательный – к анионным, если нулевой – к неэлектролитам.
Примеры:
[Cu(H2O)4]SO4, [Ni(NH3)6]Cl2, [Pt(NH3)4Br2]Cl2 – катионные комплексы,
Na3[Al(OH)6], K3[FeF6], K[Co(NH3)2(CN)4] – анионные комплексы,.
[Pt(NH3)2Cl4], [Ni(CO)4], [Cr(C6H6)2] – комплексные неэлектролиты.
б) классификация по типу лигандов
– аминокомплексы – комплексы, в которых функцию лигандов выполняют молекулы аммиака или другие амины: [Cu(NH3)4]SO4, [Pt(en)3]Cl4 (en – этилендиамин H2N(CH2)2NH2).
– аквакомплексы – комплексы, в которых функцию лигандов выполняют молекулы воды: [Cu(H2O)4]SO4, [Cr(H2O)6]Cl3, [Al(H2O)6]Cl3 .
– гидроксокомплексы – комплексы, в которых функцию лигандов выполняют гидроксид-ионы: K3[Cr(OH)6], Na2[Zn(OH)4] .
– ацидокомплексы – комплексы, в которых функцию лигандов выполняют анионы кислот: K2[PtCl6], K2[SiF6], K3[Fe(CN)6] .
Существуют также смешанные комплексы, в состав внутренней сферы которых входят различные типы лигандов.
Номенклатура комплексных соединений
Название комплексного соединения, как и название соли, начинают с названия аниона, за которым следует название катиона в родительном падеже. Для того чтобы освоить номенклатуру комплексных соединений, необходимо научиться правильно называть комплексные анионы и катионы.
Название комплексного иона (или электронейтральной комплексной частицы) начинают с указания числа лигандов и их типа. Число лигандов указывают греческими числительными:
1 – моно, 2 – ди, 3 – три, 4 – тетра, 5 – пента, 6 – гекса, 7 – гепта, 8 – окта.
Названия наиболее распространённых лигандов: F– – фторо-; Cl– – хлоро-; Br– – бромо-; I– – йодо; OH– – гидроксо-; SO32– – сульфито-; NO2– – нитро-; CN– – циано-; NCS– – родано-, NH3–аммин-; en- – этилендиамин-, H2O – аква- .
Если комплексная частица - анион, то после названия лигандов (которые перечисляют справа налево) добавляется корень латинского названия элемента-комплексообразователя и окончание «-ат».
Если в состав внутренней сферы комплекса входят в качестве лигандов и молекулы, и анионы, то в первую очередь называют анионы (с окончанием на «–о»), а затем молекулы. Если для комплексообразователя возможно несколько степеней окисления, то ее указывают в круглых скобках римской цифрой.
Примеры:
Na[Al(OH)4] – тетрагидроксоалюминат натрия
K3[Fe(CN)6] – гексацианоферрат (III) калия
K4[Fe(CN)6] – гексацианоферрат (II) калия
Na[Au(CN)2 – дицианоаурат (I) натрия
K2[Pt(NH3)2Cl4] – тетрахлородиамминплатинат (II) калия
Если комплекс катионного типа, то используют русское название (в родительном падеже) элемента-комплексообразователя:
[Ag(NH3)2]Cl – хлорид диамминсеребра
[Pt(NH3)4Cl2]Cl2 – хлорид дихлоротетраамминплатины (IV)
[Cu(H2O)4]SO4 – сульфат тетрааквамеди (II)
При составлении названия электронейтрального комплекса используют русское название комплексообразователя в именительном падеже:
[Cr(H2O)3F3] – трифторотриаквахром
[Co(NH3)3(NO2)2Cl] – хлородинитротриамминкобальт
[Pt(NH3)4Br2] – дибромотетраамминплатина
Электролитическая диссоциация комплексных соединений
Как отмечалось ранее, химическая связь между внешней и внутренней сферами комплекса – ионная, а связи центрального атома с лигандами внутренней сферы – ковалентные. Вследствие этой особенности комплексные соединения диссоциируют двояко:
На ионы внешней сферы и комплексный ион диссоциирует как сильный электролит,и такую диссоциацию называют первичнойдиссоциацией комплексного соединения:
Na2[Zn(OH)4] ® 2Na+ + [Zn(OH)4]2– .
Диссоциация внутренней сферы комплексного соединения протекает по типу слабого электролита, причем ступенчато; этот процесс называется вторичнойдиссоциацией комплексного соединения. Таким образом, вторичная диссоциация характеризуется наличием равновесия между комплексной частицей, центральным ионом и лигандами:
[Zn(OH)4]2– « [Zn(OH)3]– + OH–
[Zn(OH)3]– « Zn(OH)2 + OH–
Zn(OH)2 « ZnOH+ + OH–
ZnOH+ « Zn2+ + OH–
Диссоциация комплексных ионов, как и диссоциация других слабых электролитов, характеризуется константой равновесия. В рассматриваемом случае константа равновесия называется константой нестойкости комплексного соединения. Чем менее устойчив комплекс, тем больше его константа нестойкости. Каждой ступени вторичной диссоциации соответствует своя константой нестойкости:
[Zn(OH)4]2– « [Zn(OH)3]– + OH– 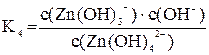
[Zn(OH)3]– « Zn(OH)2 + OH– 
Zn(OH)2 « ZnOH+ + OH– 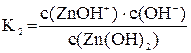
ZnOH+ « Zn2+ + OH– 
Суммарному процессу [Zn(OH)4]2– « Zn2+ + 4OH– соответствует общая константа нестойкости 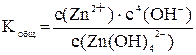 .
.
Очевидно, что Kобщ. = K1×K2×K3×K4 = К1–4
Величина, обратная константе нестойкости, называется константой устойчивости комплекса: Kобщ. уст. = 1/Kобщ. нест..
K1 уст. = 1/K1 нест. ; K2 уст. = 1/K2 нест.; K3 уст. = 1/K3 нест.; K4 уст. = 1/K4 нест. .
Чем более прочен комплекс, тем больше константа устойчивости. Константы нестойкости и устойчивости приводят в справочниках. С помощью справочных таблиц можно определить, какие комплексы наиболее характерны для заданных катионов, а также уточнить координационное число комплексообразователя (по числу ступенчатых констант).
Образование и разрушение комплексов
Аквакомплексы образуются при растворении большинства «простых» солей металлов в воде:
CuSO4 + 6H2O = [Cu(H2O)6]SO4 ;
Cu2+ + 6H2O = [Cu(H2O)6]2+ .
Соединения многих d-элементов способны взаимодействовать с аммиаком или его водным раствором с образованием амминокомплексов:
Ni(NO3)2 + 6NH3 = [Ni(NH3)6](NO3)2
Ni2+ + 6NH3 = [Ni(NH3)6]2+
Примечание: аммиачные комплексы Fe2+ и Fe3+ в водных растворах не существуют и не образуются. При действии на соли железа аммиаком выделяются малорастворимые гидроксиды. Эту особенность часто используют для отделения ионов железа от катионов других металлов, способных образовать растворимые амминокомплексы.
Гидроксокомплексы образуют амфотерные металлы:
а) CrCl3 + 6KOH = K3[Cr(OH)6]
Cr3+ + 6OH– = [Cr(OH)6]3–
б) Cr(OH)3 + 3KOHконц. = K3[Cr(OH)6];
Cr(OH)3 + 3OH– = [Cr(OH)6]3–
Ацидокомплексы можно получить в результате реакции соединений d-элементов с растворами соответствующих кислот или их солей:
а) CuSO4 + 4HClконц. = H2[CuCl4] + H2SO4
Cu2+ + 4Cl– = [CuCl4]2–
б) AgNO3 + 2KNO2 = K[Ag(NO2)2] + KNO3
Ag+ + 2 NO2– = [Ag(NO2)2]–
в) HgI2¯ + 2KIконц. = K2[HgI4]
HgI2¯ + 2I– = [HgI4]2–;
г) CuCl¯ + HCl конц. = H[CuCl2]
CuCl( + Cl– = [CuCl2]–.
Вследствие того, что комплексный ион относится к слабым электролитам, для его разрушения необходимо, чтобы в качестве продукта реакции образовался ещё более слабый (или малорастворимый) электролит. Строго говоря, большинство таких превращений относится к равновесным. Вследствие этого без количественной характеристики конкурирующих реакций (константы ионно-молекулярного равновесия) определить преимущественное направление реакции довольно сложно.
В то же время, некоторые из них протекают практически необратимо и прогнозируются на качественном уровне. Например, все гидроксокомплексы легко разрушаются при добавлении кислот, вследствие связывания лигандов (ионов OH–) с ионами H+ с образованием слабейшего электролита – H2O.
Na2[Zn(OH)4] + 2HCl. = Zn(OH)2¯ + 2NaCl + 2H2O
[Zn(OH)4]2– + 2H+= Zn(OH)2¯ + 2H2O
Na2[Zn(OH)4] + 4HСl = ZnCl2 + 2NaCl + 4H2O
[Zn(OH)4]2– + 4H+= Zn2+ + 4H2O
Большинство амминокомплексов d- металлов 4 периода разрушается при добавлении избытка кислот; при этом ионы H+ связывают молекулы NH3, образуя более слабый электролит – ионы [NH4]+ (Kнест. [NH4]+ = 5,6×10–10).
[Cu(NH3)4]SO4 + 4HCl = CuSO4 + 4NH4Cl
[Cu(NH3)4]2+ + 4H+= Cu2+ + 4NH4+
[Ag(NH3)2]Cl–+ 2HCl = AgCl¯ + 2NH4Cl
[Ag(NH3)2]+ + Cl–+ 2H+ = AgCl¯ + 2NH4+
Комплекс можно также разрушить, связав ионы внутренней сферы в малорастворимое соединение. Так, при добавлении иодида калия к раствору нитрата диамминсеребра (I) образуется осадок иодида серебра:
[Ag(NH3)2]NO3 + KI ® AgI¯ + 2NH3 + KNO3
[Ag(NH3)2]+ + I– ® AgI¯ + 2NH3
Возможно комбинированное разрушение комплекса вследствие более прочного связывания и центрального атома, и лигандов одновременно:
[Cu(NH3)4]SO4 + 3H2S ® CuS¯ + (NH4)2SO4 + 2NH4HS
[Cu(NH3)4]2+ + 3H2S ® CuS¯ + 4NH4+ + 2HS–
Количественная характеристика подобных превращений изложена в разделе «Ионно-молекулярные равновесия».
Модели химической связи в комплексных соединениях
Метод валентных связей (МВС)
В представлениях МВС ковалентные химические связи внутренней сферы, между центральным атомом и лигандами, образуются по донорно-акцепторному механизму, в результате перекрывания вакантных валентных орбиталей комплексообразователя с орбиталями лигандов, имеющих неподеленные электронные пары (рис. 9.2).


Пространственная структура внутренней сферы комплексов (МВС).
Применение МВС к прогнозированию структуры комплексов аналогично рассмотренному в разделе «Химическая связь». Для геометрии внутренней сферы комплекса определяющими оказываются валентные орбитали центрального атома (комплексообразователя).
Так, из рис. 9.2 видно, что в образовании [Ag(NH3)2]+ задействованы две орбитали Ag+, одна из них s- , другая – p-орбиталь. Для того, чтобы связи с NH3 были равными по энергии, необходима sp -гибридизация этих орбиталей, вследствие этого ион Ag(NH3)2+ имеет линейное строение. При образовании [BeF4]2– задействованы четыре вакантные орбитали цинка, одна s- типа и три – p – типа. Для равноценности образуемых связей необходима sp3 – гибридизация, вследствие этого ион должен иметь тетраэдрическую геометрию.
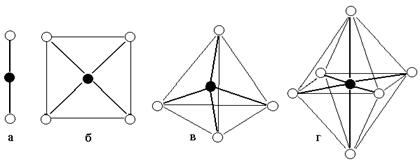

Однако есть некоторые особенности применения МВС к координационным соединениям. Например, исходя из строения валентного уровня иона Fe3+ (3d54s4p) можно предположить, что в образовании комплексов [FeF6]3– и [FeCN)6]3– будут задействованы одна s– орбиталь, три p- орбитали и две d- орбитали, тип гибридизации – sp3d2 и, соответственно, структура внутренней сферы комплексов – октаэдр. Кроме того, можно предположить, что при наличии неспаренных 3d5 электронов эти комплексы должны быть парамагнитными.
Действительно, и [FeF6]3–, и [FeCN)6]3– имеют октаэдрическую структуру и оба – парамагнитные. Однако, магнетизм [FeF6]3– соответствует наличию пяти неспаренных электронов, а [FeCN)6]3– – только одного. Как это можно объяснить в рамках МВС?
Внешне- и внутриорбитальные (высоко- и низкоспиновые) комплексы
Если в образовании связей с лигандами участвуют орбитали только внешнего валентного уровня центрального атома, то такие комплексы называют внешнеорбитальными или высокоспиновыми (cм. [FeF6]3– ).
Если в образовании связей центрального атома с лигандами участвуют кроме s-, p- орбиталей внешнего, d-орбитали предвнешнего уровня, то такие комплексы называют внутриорбитальными или низкоспиновыми.
Как может реализоваться последний вариант, если в нормальном (невозбужденном) состоянии все 3d-орбитали Fe3+ заняты электронами (в соответствии с правилом Гунда)? Можно предположить, что в комплексах, подобных [FeCN)6]3–, часть внутренних d-орбиталей могут стать вакантными в результате спаривания электронов. При этом увеличивается энергия межэлектронного отталкивания электронов, но освобождаются для образования более прочных связей орбитали, расположенные ближе к ядру (длина связи будет меньшей). По-видимому, такой вариант возбуждения Fe3+ и реализуется при образовании [FeCN)6]3–
Таким образом, изложенные выше представления МВС позволяют объяснить, но не предсказать различие в свойствах комплексов [FeF6]3– и [FeCN)6]3–. Остается открытым вопрос и о том, как связана вероятность образования внутри- или внешнесферного комплекса с особенностями электронного строения комплексообразователя и природой лигандов? Кроме того, МВС не позволяет объяснить еще одно: почему большинство соединений d-металлов окрашенные, а соединения s-, p- металлов – бесцветные?
В связи с этим потребовались дополнительные, специфические для комплексов, модели химической связи.
Теория кристаллического поля (ТКП)
В модели ТКП центральный атом рассматривается с учетом электронного строения его валентного уровня, при этом ключевое значение имеет пространственная ориентация d– орбиталей и геометрическая структура внутренней сферы комплекса. Лиганды (анионы или молекулы) рассматриваются только как точечные источники отрицательных зарядов, без учета их строения. В результате отталкивания d– электронов комплексообразователя от лигандов энергия d– орбиталей увеличивается, но, в зависимости от пространственной структуры комплекса, по-разному (рис. 9.4).
Если все лиганды рассматривать как носители отрицательного заряда, равномерно распределенного по поверхности сферы, то энергия всех d-орбиталей, независимо от их направленности, увеличится одинаково в сравнении со свободным ионом (рис. 9.4).
В реальных комплексах лиганды распределены относительно центрального атома не столь равномерно. В зависимости от структуры внутренней сферы они сосредоточены в вершинах октаэдра, тетраэдра, квадрата и др.

Рисунок 9.4..Расщепление d – орбиталей в поле лигандов различной симметрии
Октаэдрическое поле лигандов
Представим себе, что лиганды с тем же суммарным полем, что и в поле сферической симметрии, сконцентрированы в восьми симметричных точках – вершинах октаэдра. Очевидно, что при этом энергия орбиталей, направленных к вершинам октаэдра (dz2, dx2–y2), повысится по сравнению со сферическим полем, а орбиталей, направленных между осями X, Y, Z (dxy, dxz, dyz),.понизится на такую же величину (что очевидно из закона сохранения энергии) (рис. 9.4). Таким образом, d-подуровень, состоявший вначале из пяти равных по энергии орбиталей (пятикратно вырожденный) расщепился на два подуровня. Подуровень с большей энергией, состоящий из двух равных по энергии орбиталей, обозначают eg – подуровень, а подуровень с меньшей энергией, состоящий из трех равных по энергии орбиталей, обозначают t2g – подуровень. Разность энергий между подуровнями eg и t2g называется параметром расщепления d – подуровня в поле лигандов и обозначается как  или 10Dq. Величина или 10Dq зависит как от природы центрального атома, так и природы лигандов. Чем больше радиус центрального атома и чем меньше его заряд, тем больше подвержены валентные электроны полю лигандов.
или 10Dq. Величина или 10Dq зависит как от природы центрального атома, так и природы лигандов. Чем больше радиус центрального атома и чем меньше его заряд, тем больше подвержены валентные электроны полю лигандов.
В свою очередь, лиганды по силе поля располагаются в так называемый
спектрохимический ряд: I– < Br– < Cl– < OH– < F– < H2O < NH3 < NO2– < CN–
Чем правее в этом ряду расположены лиганды, тем более сильное поле онисоздают.
Из того, что суммарное повышение энергии двух орбиталей eg – подуровня должно равняться суммарному понижению энергии трех орбиталей t2g – подуровня, следует, что относительно сферического поля энергия eg - подуровня повышается на 6Dq, а энергия t2g – подуровня понижается на 4Dq.
Тетраэдрическое поле лигандов
При расположении лигандов в вершинах тетраэдра ни одна из d – орбиталей не ориентирована строго в их направлении, поэтому параметр расщепления (при том же центральном атоме и таких же лигандах) будет меньше, чем в октаэдрическом поле.
В то же время, большее влияние будет оказываться на электроны орбиталей, ориентированных между осями XYZ (dxy, dxz, dyz) и меньшее – на орбитали, ориентированные в направлении лигандов (dz2, dx2-y2). Таким образом, по сравнению с октаэдрическим полем подуровни egи t2gменяются местами.
Поле лигандов квадратной структуры
При расположении лигандов в вершинах квадрата наибольшему влиянию их поля будут подвержены электроны, расположенные на орбитали dx2-y2 (направленной к вершинам квадрата), – их энергия повысится в наибольшей мере. Повысится также (но менее, чем dx2-y2) энергия орбитали dz2 и еще менее – орбитали dxy (ориентированной между лигандами, но в плоскости XY). Понизится (в равной мере) энергия орбиталей, ориентированных в аксиальных плоскостях, т.е. dxz, dyz.
Порядок заполнение электронами орбиталей, расщепленных в поле лигандов. Высоко- и низкоспиновые комплексы.
В комплексах, образованных лигандами сильного поля, параметр расщепления (10Dq) обычно больше энергии межэлектронного отталкивания, и в таких комплексах электроны d – подуровня максимально заполняют орбитали с более низкой энергией (с соблюдением принципа Паули). В комплексах, образованных лигандами слабого поля, наоборот, параметр расщепления меньше энергии межэлектронного отталкивания, и в таких комплексах электроны d – подуровня занимают максимально возможное число орбиталей и только потом начинают спариваться. Например, nd6 – электронов в сильном октаэдрическом поле лигандов распределяются (t2g)6(eg), а в слабом поле лигандов – (t2g)3(eg)3.
Магнитные и оптические свойства комплексов. Энергия стабилизации
В зависимости от порядка заполнения d-орбиталей электронами число неспаренных электронов может быть разным. Так, в примере, рассмотренном выше, в случае лигандов сильного поля (см. [Fe(CN)6]3–) неспаренный электрон только один; а в случае лигандов слабого поля (см. [FeF)6]3–) их пять. Соответственно в первом случае магнитный момент будет
μ = √(1(1+2) = 1,7, а во втором μ = √(5(5+2) = 5,9 магнетонов Бора.
Оптические свойства и, соответственно, окраска вещества определяются величиной энергии электронных переходов с одного подуровня на другой. Различие в энергии между подуровнями s-, p-,d- достаточно велико, и поэтому поглощение энергии, связанное с таким переходами, превышает энергию, соответствующую видимой области спектра. – Вещество оказывается неокрашенным. Параметр расщепления d- подуровня в кристаллическом поле лигандов значительно меньше, поэтому электронные переходы между вновь образовавшимися d- подуровнями (например, t2g и eg в октаэдрическом поле) характеризуются энергией, соответствующей видимой области спектра. – По этой причине большинство комплексов d- металлов окрашены. В то же время, если электронная структура центрального атома d10 (например, Zn2+), то переходы электронов между d-d –орбиталями невозможны (все орбитали заняты), и такие комплексы чаще бесцветные.
Примечание: Соединения [HgI4]2– (5d10 Hg2+) и тем более [BiI4]– (висмут – p- металл) окрашены не за счет электронных переходов металла, а вследствие поляризации координированных ионов йода.
Величина изменения энергии координационного соединения в результате перераспределения d-электронов под действием поля лигандов по dε- и dγ-орбиталям называется энергией стабилизации кристаллическим полем (ЭСКП). ЭСКП рассчитывается с учетом числа d- электронов на низкоэнергетических орбиталях и орбиталях высокоэнергетических, а также с учетом распределения параметра расщепления (10 Dq) между ними.
Например, для октаэдрического высокоспинового комплекса комплекса [FeF6]3– параметр расщепления 10 Dq между подуровнями t2g (три орбитали) и eg (две орбитали) распределится следующим образом: 4 Dq – на подуровень t2g и 6 Dq на подуровень eg (в результате понижение и повышение энергии орбиталей будет равным, по 12 Dq). С учетом заполнения орбиталей электронами получим, что для трех электронов подуровня t2g суммарное понижение энергии(энергия стабилизации) будет 3*6 = 18 Dq, а для двух электронов подуровня eg энергия увеличится на 2*6 = 12 Dq (энергия дестабилизации). Таким образом, для комплекса [FeF6]3– суммарная энергия стабилизации (ЭСКП) будет 18 – 12 = 6 Dq.
Для октаэдрического комплекса [Fe(CN)6]3– параметр расщепления между подуровнями t2g и eg распределится так же: 4 Dq – на подуровень t2g и 6 Dq на подуровень eg Но, вследствие того, что этот комплекс низкосокоспиновый, и все пять электронов заполняют подуровень t2g, энергия стабилизации будет 5×6 = 30 Dq. На для подуровня eg электронов нет, поэтому энергия дестабилизации равна нулю, и ЭСКП для [Fe(CN)6]3– равна 30 Dq.
Сравнением ЭСКП однотипных комплексов можно оценить их относительную устойчивость. Из того, что ЭСКП для [Fe(CN)6]3– равна 30 Dq, а для [FeF6]3– – 0 Dq следует, что цианидный комплекс значительно прочнее фторидного (действительно, для первого К1–6 = 10–4, а для второго 10–16).
Дата добавления: 2020-07-18; просмотров: 730;
Поиск по сайту
Узнать еще

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
