КИНЕТИКА СТАЛЕПЛАВИЛЬНЫХ ПРОЦЕССОВ
Кинетика сталеплавильных процессов может быть рассмотрена как частный случай кинетики химических реакций, т. е. учения о законах их протекания во времени. В сталеплавильных агрегатах химические процессы взаимодействия осложняются протекающими одновременно процессами теплопередачи, передачи или подвода реагентов в зону реакции, искусственного или естественного перемешивания реагентов, искусственного отвода продуктов реакции и т. п. Современные представления о кинетике процесса позволяют дать ориентировочный ответ на вопрос о том, каковы скорость данного процесса и ее зависимость от отдельных параметров и стадий процесса. В общем случае процесс, протекающий в сталеплавильной ванне, может быть представлен состоящим из следующих стадий: 1) подвод реагентов к месту реакции;2) акт химической реакции; 3) выделение продуктов реакции в отдельную фазу и их удаление от места реакции.
Каждую из стадий можно подразделить на ряд промежуточных. Скорость процесса в целом vr зависит от скорости протекания каждой стадии: v,,v2, v3, ...и т. д., т.е.
V∑=1/(1 /V1+ 1/V2+1/V3 + ...).
В каждом конкретном случае любая из этих стадий может лимитировать процесс в целом, если скорость ее протекания меньше, чем других. Если, например, скорость V2 снизится до нуля, то значение l/V2 возрастает до 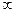 , a V
, a V 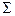 уменьшается до нуля.
уменьшается до нуля.
8.2.1. Скорость протекания реакции.В основе кинетики химических реакций как учения о скоростях химических превращений лежит закон действующих масс, согласно которому скорость реакции веществ А, В, С,... пропорциональна произведению их концентраций. Скорость реакции обычно характеризуется уменьшением за единицу времени концентрации какого-либо из исходных веществ или конечных продуктов реакции. Например, скорость вступления в реакцию вещества А (скорость уменьшения его концентрации в единицу времени) выражается уравнением
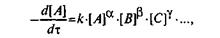
где k —константа скорости реакции; [А], [В], 80
[С],... — концентрации взаимодействующих веществ; минус показывает, что концентоа-ция вещества А убывает со временем. Сумму величин 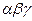 называют порядком реакции.
называют порядком реакции.
Константа скорости реакции численно равна скорости реакции, если концентрация каждого из исходных веществ равна единице. Практически значения скоростей реакций, протекающих в реальных сталеплавильных процессах, значительно отличаются от определенных в соответствии с законом действующих масс, так как, во-первых, приходится иметь дело с растворами того или иного компонента в шлаке или металле и учитывать влияние растворителя (т. е. неидеальность раствора); во-вторых, реакции в реальных условиях протекают, как правило, в гетерогенных, а не в гомогенных средах, в связи с чем скорости реакций часто определяют для каждого конкретного случая экспериментально. Обычно скорость реакции выражается в изменении концентрации вещества в процентах от массы металла (или шлака) в единицу времени (час или минута); например, скорость окисления углерода составляет -0,5 % С/ч и т. п.
8.2.2. Энергия активации: понятие об активированном комплексе.Влияние температуры на константу скорости химической реакции k видно из уравнения Аррениуса1
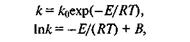
где kf, — константа (так называемый предэкс-поненциальный множитель константы скорости); Е— энергия активации реакции; R — универсальная газовая постоянная.
Энергию активации Е обычно определяют экспериментально следующим образом: исследуют кинетику реакции при нескольких температурах и строят график (рис. 8.2) в координатах Ink— 1/T тангенс угла наклона прямой 1 на этом графике в соответствии с уравнением Аррениуса равен Е. В более сложных случаях зависимость Ink от величины 1/T выражается кривой 2
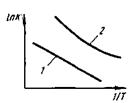
Рис. 8.2.Зависимость константы скорости реакции от температуры
Константа скорости реакции k, а следовательно, и скорость реакции значительнее изменяются с изменением температуры в тех реакциях, в которых энергия активации больше.
Физический смысл энергии активации сводится к следующему: в реакциях, протекающих с конечной скоростью, число столкновений между молекулами, приводящих к химическому взаимодействию, составляет лишь некоторую (небольшую) часть от общего числа столкновений. Эффективными оказываются лишь столкновения между такими молекулами, между такими конфигурациями атомов, которые в момент столкновения обладают некоторым избытком внутренней энергии по сравнению со средней энергией частиц при данной температуре. Этот избыток энергии определяется энергией активации.
На рис. 8.3 по оси ординат отложена энергия рассматриваемой системы молекул, а по оси абсцисс — ход реакции (в единицах времени или массы прореагировавшего вещества). Если прямая реакция (переход из состояния / в состояние II) является эндотермической, то общий запас энергии продуктов реакции меньше энергии исходных веществ, т. е. система в результате этой реакции переходит на более низкий энергетический уровень II. Разность энергий уровней / и // определяет тепловой эффект реакции Q. Уровень М соответствует тому наименьшему запасу энергии, которым должны обладать молекулы, чтобы их столкновения могли приводить к химическому взаимодействию. Разность энергий уровней М и I представляет энергию активации прямой реакции E1, а разность энергий уровней М и // — энергию активации обратной реакции ei. Таким образом, при переходе из исходного состояния в конечноесистема должна преодолеть своего рода энергетический барьер. Иногда помимо истинной энергии активации, определяемой по уравнению Аррениуса, используют понятие кажущаяся энергия активации. Кажущаяся энергия активации может отличаться от истинной, например, на величину тепловых эффектов, сопровождающих процессы адсорбции и десорбции взаимодействующих веществ, и т. п. Другими словами, величина кажущейся энергии активации не учитывает влияние всех факторов на кинетику процесса.
В теории скоростей химических реакций широко используют понятие активированный комплекс — это группировка атомов в решающий момент элементарного акта химической реакции.
В ходе элементарного акта реакции возникает состояние, являющееся критическим в том смысле, что если оно достигнуто, то дальнейшее движение атомов происходит беспрепятственно, не требуя запаса энергии. Совокупность атомов в этом состоянии принято называть активированным комплексом или переходным состоянием. Если ввести понятие концентрация активированного комплекса прямой реакции Сакт и обозначить продолжительность жизни этих комплексов т, то скорость прямой реакции v представляет собой число соответствующих актов реакции в единице объема в единицу времени, т. е. v = Сакт  . Величины Сакт и вычисляют методами статистической механики, а их соотношение позволяет устанавливать абсолютное значение скорости реакции. Если принять представление об активированных комплексах, то энергия активации равна разности значений средних энергий активированных комплексов и исходных молекул.
. Величины Сакт и вычисляют методами статистической механики, а их соотношение позволяет устанавливать абсолютное значение скорости реакции. Если принять представление об активированных комплексах, то энергия активации равна разности значений средних энергий активированных комплексов и исходных молекул.
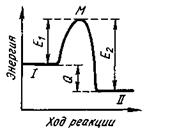
Рис. 8.3.Схема изменения энергии системы в течение реакции
Для случая, когда число активных молекул Na составляет относительно малую долю от общего числа молекул N, их отношение может быть представлено уравнением Больцмана1
Na/ N= ехр(-E/RT);
lnNaa/ N= -E/RT,
где Na/ N — относительное число активных молекул.
Зная энергию активации, можно определить относительное число активных молекул. Если величина Na/N значительно меньше 10-20, то скорость реакции чрезвычайно мала и реакция практически не идет. Если Na/ N существенно больше 10-10, то реакция в большинстве случаев происходит практически мгновенно. Значениям Na/ N от 10-20 до 10-10 соответствует величина Е/Т в пределах от 100 до 50, что для температур сталеварения (порядка 2000 К) соответствует значениям энергии активации от 840 до 420 кДж. По имеющимся данным, значения энергии активации большинства реакций сталеплавильных процессов существенно ниже указанных значений, т. е. акт химического взаимодействия не должен быть лимитирующим для процесса в целом.
8.2.3. Условия зарождения новой фазы.В тех случаях, когда в пределах исходной фазы должна образоваться новая фаза (например, пузырь газа внутри стальной ванны, неметаллическое включение, твердый кристалл металла и т. п.), скорость процесса при прочих равных условиях (давление, температура и т. п.) может лимитировать третья стадия процесса: зарождение и выделение новой фазы. Это связано с тем, что в обычных условиях зародыши любой новой фазы становятся устойчивыми лишь после достижения определенных значительных размеров, а рост зародыша до этих размеров приводит не к уменьшению, а к увеличению энергии Гиббса системы (образование зародыша новой фазы внутри исходной приводит/к созданию поверхности раздела фаз/ а это, в свою очередь, связано с затратой энергии).
1 L. Boltzmann (1844— 1906) — австрийский физик, основатель статистической физики и физической кинетики.
Общее изменение энергии Гиббса при протекании процесса ΔGпроц;складывается в данном случае из изменения энергии Гиббса при протекании химической реакции ΔGхр и образовании поверхности новой фазы ΔGпов
:
ΔGпроц;= ΔGхр+ ΔGпов
где Δ G ПОВ= f(r зар)  — функция размеров зародыша (гзар — радиус зародыша, так как обычно принимают, что зародыш имеет сферическую форму).
— функция размеров зародыша (гзар — радиус зародыша, так как обычно принимают, что зародыш имеет сферическую форму).
Рост зародыша до известных пределов связан с затратой энергии, т. е. свободная энергия системы возрастает. По достижении зародышем определенного размера, называемого критическим, изменение энергии Гиббса достигает максимума; при дальнейшем увеличении зародыша энергия Гиббса системы уменьшается и процесс начинает протекать самопроизвольно.
Зародыш новой фазы, имеющий критические размеры, находится в состоянии неустойчивого равновесия с окружающей его средой: если размеры его увеличатся, он будет самопроизвольно расти дальше и суммарная величина Δ G проц будет уменьшаться; если же размер зародыша немного уменьшится, то последует дальнейшее самопроизвольное его уменьшение, вплоть до полного исчезновения. В соответствии с этим зародыш критического размера называют иногда также равновесным. Значение энергии Гиббса системы до начала процесса G1 больше, чем после завершения процесса G2. Таким образом, величина Δ G пр0ц= G2 - G1является отрицательной. При этом слагаемыми ΔGnpou в формуле ΔGnpou = ΔGХр + ΔGnoв являются: ΔG хр — зависит от количества (объема) вещества, т. е. величина, пропорциональная г3 зар, и ΔGпов—зависит от поверхности (площади) новой фазы, т. е. величина, пропорциональная г2 зар. При очень малых начальных значениях радиуса зародыша г зар величина г3 зар значительно меньше г2 зар (г3 зар < г2 зар ) и роль ΔGnов будет определяющей. Для случая зародыша сферической формы ΔGnов = 4 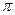 r2зар
r2зар 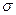 . Чем меньше поверхностное натяжение а, тем меньшая энергия затрачивается на образование поверхности, однако во всех случаях при малых значениях гзар величина ΔGnов определяет возможность протекания процесса.
. Чем меньше поверхностное натяжение а, тем меньшая энергия затрачивается на образование поверхности, однако во всех случаях при малых значениях гзар величина ΔGnов определяет возможность протекания процесса.
Самопроизвольное образование зародышей новой фазы внутри исходной связано с явлениями флуктуации1. В результате флуктуации в веществе могут возникать разные сочетания скоплений молекул или атомов, имеющие размеры, достаточные, чтобы служить центрами для выделения новой фазы в данных условиях. Вероятность возникновения зародышей новой фазы зависит от степени пересыщения раствора и от других факторов. Чем выше степень пересыщения (при данной температуре), тем выше вероятность возникновения зародышей. Размер критического радиуса зародыша rзаркрит прямо пропорционален поверхностному натяжению и обратно пропорционален степени пересыщения. Чем выше степень пересыщения
и чем меньше , тем меньше rзаркрит .
В случае, когда новая фаза уже образовалась, изменение ΔGnов зависит от поверхностного натяжения и от характеристик состояния фазы, в которую переходят продукты реакции:
ΔGnов = 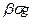 -1/3,
-1/3,
где ( 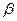 — коэффициент, зависящий от молекулярной массы, плотности и геометрической формы фазы, в которую переходят продукты реакции; g— общее число молекул в фазе, в которую переходят продукты реакции.
— коэффициент, зависящий от молекулярной массы, плотности и геометрической формы фазы, в которую переходят продукты реакции; g— общее число молекул в фазе, в которую переходят продукты реакции.
На поверхности раздела фаз значение g очень велико и ΔGnов = 0, поэтому наличие готовых поверхностей раздела существенно ускоряет процессы образования новой фазы. Такими поверхностями раздела могут быть, например, границы: металл—футеровка, металл—пузырь газа, металл—взвешенные твердые частички включений и т.д.
8.2.4. Роль диффузии.В большинстве случаев сталеплавильная ванна представляет собой систему, в которой поверхности раздела фаз уже имеются и они достаточно велики (например, при интенсивной продувке ванны), т. е. процессы выделения новой фазы существенно облегчены. В этих случаях, как уже отмечалось, скорость процесса в целом определяется скоростями массопереноса.
Напомним, что процессы перемещения компонента внутри фазы обычно называют массопереносом, а через границу раздела фаз — массопередачей. В отдельные периоды конвертерной плавки скорость окисления углерода зависит от того, с какой интенсивностью вдувается через фурму газообразный кислород в ванну. Другими словами, скорость процесса обезуглероживания определяется массопереносом кислорода.
Диффузия2 — движение частиц среды, приводящее к переносу вещества и к выравниванию концентраций. В процессе диффузии возможно "взаимное проникновение веществ вследствие теплового движения их частиц. В газах диффузия происходит очень быстро, в жидкостях — медленнее, в твердых телах — весьма медленно. В работах физика-теоретика А. Эйнштейна показано, что средний квадрат смещения частиц при диффузии пропорционален продолжительности диффузии. Коэффициент пропорциональности этого соотношения обозначается обычно латинской буквой D и называется коэффициентом диффузии. При повышении температуры значения D возрастают. Это важно помнить.
Если, например, реакция удаления серы из металла в шлак протекает на границе металл—шлак, то чем выше температура, тем более интенсивно идет диффузия серы из объема металла к этой границе, тем скорее удаляется сера из металла. Или, например, если коэффициент диффузии водорода в жидком железе DH = 8 • 10 -3 см2/с, а азота dn = 4 • 10 -5 см2/с, то это значит, что скорость диффузии водорода во много раз больше, чем азота.
Немецкий ученый А. Фик в 1855 г. сформулировал закон (I закон Фика), в соответствии с которым диффузионный поток пропорционален градиенту концентраций диффундирующих компонентов (на единицу длины). Диффузионным потоком принято называть поток массы, диффундирующий через единицу площади в единицу времени (размерность — кг/(м2 -с)). При рассмотрении процессов диффузии в жидкой ванне (металле и шлаке) важно учитывать сильное влияние вязкости ( 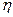 ). Напомним, что D- = const, т.е. чем больше вязкость, тем слабее проявляются процессы диффузии.
). Напомним, что D- = const, т.е. чем больше вязкость, тем слабее проявляются процессы диффузии.
1 Флуктуации (от лат. fluctuatio — колебание) — случайные отклонения наблюдаемых величин от их средних значений.
2 От лат. diffusio — распространение, растекание, рассеивание.
Дата добавления: 2016-06-22; просмотров: 2618;
Поиск по сайту
Узнать еще

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
