Ихтиоз (ichthyosis)
Ихтиоз (син.: диффузная кератома, сауриаз, кожа аллигатора) — наследственное заболевание, характеризующееся диффузным нарушением ороговения по типу гиперкератоза.
Этиология заболевания неизвестна. Заболевание обусловлено мутантными генами, биохимический эффект которых еще не расшифрован. При ихтиозе отмечены нарушения липидного, белкового, в частности аминокислотного, обмена [Кряжева С. С., 1977], усиление активности окислительных ферментов эпидермиса [Шмыгло М. П., 1972, и др.]. Определенное значение имеют недостаточность витамина А, эндокринопатии (недостаточность функции щитовидной железы и др.), недостаточность клеточного и гуморального иммунитета [Кряжева С. С. и др., 1979]. Группа ихтиоза гетерогенна.
Различают следующие разновидности ихтиоза: вульгарный, сцепленный с полом (Х-хромосомой), ламеллярный, эпидермолитический, плода, односторонний, линеарный огибающий, иглистый. Ряд синдромов включают ихтиоз как один из симптомов: синдром Нетертона, синдром Рефсума, синдром Руда, синдром Шегрена—Ларссона, синдром Юнга—Фогеля.
Ихтиозиформные изменения кожи могут быть непостоянным симптомом при таких наследственных заболеваниях, как врожденная кальцифицирующая хондродисплазия, синдром Конради— Хюнерманна, синдром мальабсорбции.
Наследственные формы ихтиоза (истинный ихтиоз) следует дифференцировать от следующих приобретенных заболеваний: приобретенного ихтиоза (старческий, симптоматический, дисковидный) и эритррдермий (эксфолиативный дерматит Риттера, эксфолиативная эритродермия Лейнера, эритродермическая форма красного отрубевидного волосяного лишая).
Вульгарный ихтиоз (ichthyosis vulgaris) наследуется по аутосомно-доминантному типу. Проявляется в первые месяцы или (реже) годы жизни. Кожа туловища, конечностей (за исключением складок, ладоней, подошв) становится сухой, шершавой, грязно-серого цвета с большим количеством чешуек. В зависимости от цвета чешуек, характера их прикрепления к подлежащей коже и степени их выраженности различают ксеродерму, простой, белый, блестящий и фолликулярный ихтиоз. Кожа лица поражается обычно у взрослых. У детей изменения кожи отсутствуют или выражены незначительно. На коже ладоней отмечается четкий рисунок кожных линий в виде мелкой складчатости кожи. Ногтевые пластины ломкие, сухие, возможны деформации ногтей. Сало- и потоотделение снижено.
Заболевание длится всю жизнь. Клинические проявления его ослабевают в период полового созревания и в зрелом возрасте. Вульгарный Ихтиоз нередко сочетается с нейродермитом [Korting С. W. et al., 1967], себорейной экземой, крипторхизмом, осложняется пиодермией.
При гистологическом исследовании выявляются ретенционный гиперкератоз — средний гиперкератоз с истончением или полным отсутствием зернистого слоя. Шиповатый слой не изменен или слегка редуцирован. Сосочковый слой дермы нормальный. Волосяные фолликулы и сальные железы несколько редуцированы, отмечается периваскулярная инфильтрация дермы.
При электронно-микроскопическом исследовании выявлены низкая дифференциация тонофиламентов, редукция и аномалия кератогиалинового синтеза в сочетании с усилением рибосомальной активности [Schnyder U. W., 1975]. Пролиферативная активность в эпидермисе при вульгарном ихтиозе нормальная, наблюдается нарушение отшелушивания рогового слоя [Frost P., 1973].
Гистохимические исследования [Куклин В. Т., 1971] позволили выявить при вульгарном ихтиозе уменьшение ядер и количества ДНК в клетках базального и шиповатого слоев, пиронинофилию цитоплазмы клеток шиповатого слоя, обусловленную нарушением белкового, в частности аминокислотного, обмена. В роговом слое увеличено содержание аминогрупп, триптофана, SH-групп и гликомукопротеидов. Замедленное отшелушивание рогового слоя, по-видимому, объясняется увеличением содержания гликозаминогликанов и их цементирующим действием.
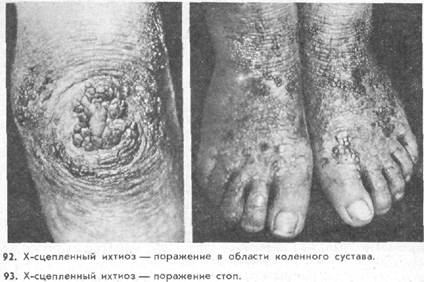
Ихтиоз, сцепленный с полом, выделен Е, A. Cockayne (1933) на основании результатов генетического исследования как самостоятельная нозологическая единица. Генетические, клинические и гистологические особенности этой формы описаны R. S. Wells и С. В. Кегг (1965). Болеют только мужчины. Заболевание наследуется рецессивно, сцеплено с Х-хромосомой. Клинические признаки ихтиоза развиваются через несколько недель после рождения. Чаще поражен весь кожный покров, включая области сгибов (у 30% больных). У детей в процесс всегда включаются волосистая часть головы, лицо, шея. Ладони и подошвы не поражены. В отличие от вульгарного ихтиоза чешуйки большие, темные, часто формируются щитки, массивные роговые наслоения типа панциря ящерицы, игл ежа (рис. 92, 93). Для этой формы характерно также поражение глаз в виде катаракты [Sever R.I. et al., 1968]. В редких случаях могут наблюдаться умственная отсталость, эпилепсия, гипогонадизм, у больных отмечен дефицит стероидфосфатазы.
При гистологическом исследовании выявляют гиперкератоз, гранулез, небольшой акантоз. Выражены гипертрофия сосочкового слоя дермы, периваскулярные инфильтраты. Ряд авторов [Frost P., 1973, и др.] считают, что при этой форме ихтиоза пролиферативная активность эпидермиса нормальная, нарушены процессы отшелушивания рогового слоя. R. Schwarz и соавт. (1969) отмечали при этой форме ихтиоза в отличие от других форм выраженное уменьшение содержания серина в чешуйках кожи.
Дифференциальная диагностика. Вульгарный и X-сцепленный ихтиоз дифференцируют от приобретенного ихтиоза и ламеллярного ихтиоза.
Приобретенный ихтиоз в отличие от вульгарного и Х-сцепленного ихтиоза обычно развивается в более позднем возрасте, имеет симптоматический характер, проявляясь при гиповитаминозе А, злокачественных новообразованиях внутренних органов и др. Приобретенный ихтиоз как паранеопластический процесс наблюдается при болезни Ходжкина, висцеральном раке, грибовидном микозе, хотя и встречается довольно редко. Клиническая картина напоминает таковую при вульгарном ихтиозе, но поражаются также кожные складки, ладони и подошвы. Больных беспокоит зуд, который обычно предшествует изменениям кожи. При электронно-микроскопическом исследовании выявляют небольшие керато-гиалиновые гранулы нормальной структуры, кератосомы также не изменены в отличие от вульгарного ихтиоза.
Возрастные изменения кожи (сенильный ихтиоз) характеризуются резкой сухостью кожи, образованием трещин, крупнопластинчатым шелушением, развивающимся в пожилом возрасте. Наблюдаются также другие признаки инволюции кожи: истончение, потеря эластичности, побледнение, дисхромия, дистрофия придатков кожи — волос, ногтей, сальных и потовых желез. Нередко образуются старческие кератомы.
Ихтиоз плода (ichthyosis faetalis) — врожденный ихтиоз, развивается в эмбриональном периоде (на IV—V месяце беременности). Заболевание наследуется по аутосомно-рецессивному типу. К моменту рождения клиническая картина ихтиоза полностью сформирована. Кожа новорожденного покрыта роговым панцирем, состоящим из толстых роговых щитков серо-черного цвета, толщиной до 1 см, гладких или зазубренных, разделенных бороздами и трещинами. Ротовое отверстие нередко растянуто, малоподвижно или, наоборот, резко сужено, напоминает хобот, едва проходимо для зонда. Нос и ушные раковины деформированы, веки выворочены. Конечности уродливые (косорукость, контрактуры, аингум). Волосы и ногти отсутствуют или дистрофичны. Большинство детей рождаются мертвыми или умирают вскоре после рождения от истощения и сепсиса.
При гистологическом исследовании выявляют выраженный гиперкератоз. Зернистый слой атрофичен, в сосочковом слое дермы лимфоцитарные инфильтраты.
 К врожденному ихтиозу относят также ихтиозиформную врожденную эритродермию Брока (erythrodermia ichthyosiformis congenita Brocq). Различают небуллезную и буллезную форму ихстиозиформной эритродермии. Небуллезную разновидность многие авторы считают тождественной ламеллярному ихтиозу. Буллезную разновидность чаще называют «эпидермолитический гиперкератоз», этот термин широко распространен в литературе,
К врожденному ихтиозу относят также ихтиозиформную врожденную эритродермию Брока (erythrodermia ichthyosiformis congenita Brocq). Различают небуллезную и буллезную форму ихстиозиформной эритродермии. Небуллезную разновидность многие авторы считают тождественной ламеллярному ихтиозу. Буллезную разновидность чаще называют «эпидермолитический гиперкератоз», этот термин широко распространен в литературе,
Ламеллярный ихтиоз [ichthyosis lamellaris] наследуется по аутос ом но-ре цессив ному типу. Заболевание проявляется при рождении картиной так называемого коллоидального плода.
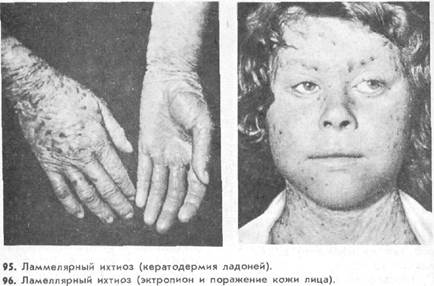
Однако «коллоидальный плод» может быть начальным проявлением не только ламеллярного ихтиоза, но и других форм — ихтиоза, сцепленного с полом, эпидермолитического ихтиоза, синдрома Шегрена - Ларсона и др. [Reed W., 1972]. Покрасневшая кожа ребенка при рождении полностью покрыта тонкой сухой желтовато-коричневой пленкой, напоминающей коллодий. В части случаев коллодийная пленка, просуществовав некоторое время, превращается в крупные чешуйки (ламмелярная эксфолиация новорожденных) и еще в раннем грудном возрасте полностью исчезает и кожа на протяжении всей жизни остается нормальной. В большинстве же случаев чешуйки, образующиеся из пленки, остаются на всю жизнь — ламеллярный ихтиоз (рис. 94).
С возрастом эритродермия регрессирует, а гиперкератоз усиливается. Поражение распространяется на все кожные складки, причем изменения кожи в них часто более выражены. Кожа лица обычно красная, натянутая, шелушится. Волосистая часть головы покрыта обильными чешуйками. Наблюдается повышенное потоотделение на ладонях, подошвах, лице и других участках кожи. Волосы и ногти растут быстро (гипердермотрофия), ногтевые полностью исчезает и кожа на протяжении всей жизни остается гиперкератоз, диффузный кератоз ладоней (рис. 95) и подошв. Характерным проявлением ламеллярного ихтиоза является также эктропион (рис. 96), которому нередко сопутствуют лагофтальм, кератит, фотофобия [Sever R. J. et al., 1968]. Иногда при ламеллярном ихтиозе наблюдается умственная отсталость.
При гистологическом исследовании выявляют пролиферационный гиперкератоз (иногда с паракератозом), гранулез, умеренный акантоз, гипертрофию сосочков дермы, увеличение сальных и потовых желез, периваскулярные инфильтраты.
При электронно-микроскопическом исследовании отмечены увеличение количества кератиносом в верхних отделах шиповатого слоя, плохое контурирование тонофиламентов, большое количество митохондрий в зернистом слое, что свидетельствует о возросшем эпидермопоэзе в сочетании с увеличением сцепления роговых клеток [Vandesteen P. R., Muller S. А. 1972]. Гистохимически обнаружено увеличение окислительных ферментов в эпидермисе.
Дифференциальная диагностика. Заболевание следует дифференцировать от вульгарного и Х-сцепленного с полом ихтиоза, десквамативной эритродермии Лейнера — Муссу, эксфолиативной эритродермии Вильсона — Брока, синдрома Нетертона, псориатическои эритродермии, эритродермической формы красного отрубевидного волосяного лишая, эритрокератодермии.
Вульгарный и сцепленный с полом ихтиоз отличаются от ламеллярного ихтиоза тем, что развиваются несколько позднее (в течение первых недель, месяцев жизни), не являясь врожденной патологией, как ламеллярный ихтиоз. В отличие от ламеллярного ихтиоза при этих формах не наблюдаются воспалительный фон (эритродермия), кератодермия, эктропион, гипер-дермотрофия и такая выраженная дистрофия ногтей с подногтевым гиперкератозом и утолщением ногтевых пластин.
В детском возрасте определенные трудности возникают при дифференциальной диагностике ламеллярного ихтиоза с десквамативной эритродермией Лейнера — Муссу. При этом следует иметь в виду, что последняя развивается в первые недели жизни ребенка (не являясь врожденной) у детей с гипотрофией, у которых имеется дефицит витаминов комплекса В. Поражение начинается с образования участков опрелости кожи (чаще в аногенитальной области), лишь в дальнейшем (в отличие от ламеллярного ихтиоза) сливающихся в сплошную эритродермию с десквамацией эпидермиса. Кожа ладоней и подошв напряжена, красная, блестящая в отличие от утолщенной по типу диффузного кератоза кожи ладоней и подошв при ламеллярном ихтиозе. Характерны также расстройства деятельности желудочно-кишечного тракта (понос, срыгивания), изменения крови — гипохромная анемия, лейкоцитоз, что обычно не наблюдается при ламеллярном ихтиозе. Заболевание длится до 1 мес, после чего все проявления его исчезают в отличие от ламеллярного ихтиоза, сохраняющегося в течение всей жизни.
У взрослых ламеллярный ихтиоз следует дифференцировать от эритродермии Вильсона — Брока и эритродермий — проявлений гемобластозов (грибовидный микоз и др.). В сформированной клинической картине при этих состояниях, так же как при ламеллярном ихтиозе, отмечается эритродермия с крупнопластинчатым шелушением (однако чешуйки не прикрепляются так плотно и не имеют коричневато-серого оттенка, как при ламеллярном ихтиозе), кератодермия, дистрофия ногтей с подногтевым гиперкератозом. Основным ориентиром в диагностике могут служить позднее начало заболевания (в отличие от врожденного характера ламеллярного ихтиоза), нередко постепенное развитие эритродермии, начиная с отдельных зудящих эритемато-сквамозных пятен, полиаденит и специфический характер инфильтрата при гемобластозах (см. их отдельные нозологические формы).
Синдром Нетертона включает ламеллярный ихтиоз как один из симптомов, однако синдром отличает наличие проявлений атопии (бронхиальная астма, аллергический ринит, нейродермит и др.) и своеобразной аномалии волос (перекрученные волосы, инвагинирующий трихорексис и др.). Кроме того, необходимо иметь в виду, что при синдроме Нетертона вместо ламеллярного ихтиоза может наблюдаться поражение кожи в виде огибающего линеарного ихтиоза.
Эпидермолитический ихтиоз (ichthyosis ephidermolitica) — редкая форма врожденного ихтиоза, наследуемая аутосомно-доминантно. Заболевание проявляется сразу после рождения в виде «ошпаренной кожи». Кожа новорожденного ярко-красного цвета, имеются обширные участки отслоения эпидермиса, с образованием эрозий и пузырей различной величины с вялой покрышкой и положительным симптомом Никольского (симптом отслойки пузыря). Кожа ладоней и подошв утолщена, беловатого цвета, эктропиона нет. В тяжелых случаях процесс сопровождается геморрагическим компонентом (пурпура) и приводит к летальному исходу. В более легких случаях дети выживают. Чаще с возрастом количество пузырей резко уменьшается, а ороговение кожи усиливается неравномерно на разных участках. На 3—4-м году жизни отчетливо выявляется гиперкератоз. Клиническая картина напоминает таковую при ламеллярном ихтиозе, но лицо обычно не поражено, за исключением легкого кератоза носогубных складок, волосы и ногти нормальные. На коже туловища может наблюдаться гиперкератоз типа hystrix, почти генерализованный, но неравномерный, сильнее выраженный в области складок кожи, где особенно хорошо видна поперечная складчатость. Периодически на коже появляются пузыри и эрозии.
При гистологическом исследовании выявляют акантокератолитический гиперкератоз, роговой слой утолщен, с участками паракератоза, отмечаются гранулез, акантоз, признаки дискератоза-круглые тельца и зерна. Базальный слой сформирован хорошо. В шиповатом слое межклеточный отек, в средних и верхних отделах клетки шиповатого слоя содержат мелкие и крупные базофильные гранулы и пикнотические ядра, границы клеток нечеткие. Сосочки дермы гипертрофированы. В верхних слоях дермы выраженный воспалительный инфильтрат.
При электронной микроскопии выявляют вертикальную ориентацию кератиноцитов в роговом слое, в результате чего за счет атипичного рогового слоя повышаются трансэпидермальная потеря воды [Frost P., 1968) и топическая кортикостероидная адсорбция. В роговом слое обнаруживают также признаки неполной кератинизации клеток, в зернистом слое большое число клеток с выраженной вокруг ядерной зоной, содержащей базофильный кератогиалин, митохондрии, кератиносомы, скопления кератиносом в межклеточных пространствах зернистого и рогового слоев. Тонофиламенты шиповатого слоя утолщены в виде широких прядей, глыб, околоядерных скоплений, размер и количество керато-гиалиновых глыбок увеличены. Комплекс десмосомы — тонофиламенты не нарушен (в отличие от акантолитических дерматозов — вульгарной пузырчатки, болезни Дарье и др.).
Дифференциальная диагностика. Эпидермолитический ихтиоз следует отличать от эксфолиативного дерматита Риттера и буллезного эпидермолиза.
Эксфолиативный дерматит Риттера может напоминать эпидермолитический ихтиоз у детей раннего возраста, так как в этот период еще резко выражен эритродермический компонент. Необходимо иметь в виду, что в отличие от эпидермолитического ихтиоза ребенок рождается с нормальной кожей. Заболевание развивается в первые дни жизни (в конце 1-й — начале 2-й недели), протекает остро (тяжелое общее состояние, высокая температура тела). Первые очаги поражения кожи возникают на лице, вокруг рта в виде рожеподобного покраснения кожи, что также не характерно для ламеллярного ихтиоза. Через несколько дней эритема захватывает всю голову, туловище, конечности (эритематозная стадия), появляются мокнутие, трещины и отслойка эпидермиса (положительный симптом Никольского) в виде широких пластов (эксфолиативная стадия), обнажается вишневого цвета дерма. Настоящие же пузыри образуются редко. Через несколько дней процесс стихает (регенеративная стадия). На коже ладоней и подошв происходит отслойка эпидермиса.
Дифференциальную диагностику с буллезнымэпидермолизом, для которого также характерно образование пузырей, проводят в ряде случаев в ранних стадиях заболевания на основании отсутствия диффузных воспалительных изменений кожи (эритродермия) и обширного слущивания эпидермиса при буллезном эпидермолизе в отличие от эпидермолитического ихтиоза. В связи с усилением в дальнейшем гиперкератоза при ихтиозе различие этих заболеваний становится еще более существенным.
Односторонний ихтиоз(ichthyosis unilateralis; син.: односторонняя врожденная ихтиозиформная эритродермия Россмана) многие авторы рссматривают как атипичный вариант эпидермолитического ихтиоза. Тип наследования заболевания неизвестен. Патологический процесс односторонний (слева или справа), захватывает половину лица, туловища, верхнюю и нижнюю конечности. Обычно сочетается с костными деформациями, кистозными изменениями почки (на той же стороне), мозговыми нарушениями, которые обнаруживают с помощью электроэнцефалографии [CullenS. J., 1969].
Дифференциальная диагностика не представляет затруднений в связи с выраженным односторонним характером поражения в сочетании с костной и висцеральной патологией, не наблюдающимся ни при одном другом виде ихтиоза.
Линеарный огибающий ихтиоз Комеля (ichthyosis linearis cir-cumflexa Cornel). Принадлежность этой нозологической формы к ихтиозу окончательно не выяснена. Н. В. Литвинок и 3. М. Тузова (1966) и другие авторы считают ее вариантом ихтиозиформной эритродермии, хотя некоторые предполагают, что это разновидность вариабельной эритрокератодермии. Заболевание возникает в первые дни жизни. Высыпания обычно локализуются на туловище, сгибательных поверхностях конечностей и состоят из полициклических или кольцевидных серпигинозных участков эритемы, нередко с сероватым оттенком, окруженных чуть приподнятым слабо-розовым валиком с пластинчатым шелушением и небольшим количеством мелких, быстро ссыхающихся пузырьков, расположенных под роговым слоем. Вследствие изменчивости очертаний создается впечатление роста очагов поражения. Кожа на локтевых сгибах и в подколенных впадинах лихенизирована. На волосистой части головы отмечается мелкопластинчатое шелушение. Наблюдается локализованный ладонно-подошвенный гипергидроз. Ногти и слизистые оболочки не изменены. Течение заболевания хроническое, с периодическими самопроизвольными ремиссиями. В некоторых случаях наблюдается резкое обострение заболевания с развитием универсальной эритродермии, сопровождающейся лихорадкой, ухудшением общего состояния больного. Иногда наблюдается отставание в психическом развитии. При гистологическом исследовании выявляют очаговый паракератоз, гранулез, субкорнеальные полости, умеренный акантоз, небольшой отек дермы с лимфогистиоцитарными инфильтратами вокруг сосудов.
Дифференциальная диагностика. Линеарный огибающий ихтиоз Комеля дифференцируют от дисковидного ихтиоза, синдрома Нетертона, эритрокератодермий.
Дисковидный ихтиоз (ichthyosis discoides; син.: кольцевидный лишай) — заболевание неизвестного генеза, развивается в детском возрасте, обычно после тяжелых болезней, характеризуется обрат зованием участка инфильтрации кожи за счет слияния кольцевидных очагов гиперкератоза [Radhakrishnamurthy К., Ratnakumari С., 1973].
Синдром Нетертона может включать линеарный ихтиоз как один из симптомов. Кроме линеарного ихтиоза, отмечаются аномалии волос и аллергический статус в виде бронхиальной астмы, нейродермита и др.
Иглистый ихтиоз (ichthyosis hystrix) — редкая форма ихтиоза, наследуемая аутосомно-доминантно. Термин «ichthyosis hystrix» применяли исторически для обозначения различных веррукозных линеарных состояний и поэтому в качестве синонимов использовали термины «эпидермальный невус», «буллезный ихтиозиформный гиперкератоз» и др. Путаница в терминологии объясняется клиническим сходством аутосомно-доминантного заболевания ichthyosis hystrix и веррукозного эпидермального невуса.
При рождении заболевание проявляется лишь выраженной эритемой без шелушения и пузырей. В течение нескольких недель эритема ослабевает, появляется диффузное шелушение, азатем образуются линеарные зоны массивных веррукозных роговых наложений, напоминающих иглы ежа. Участки усиленного образования рогового слоя имеют V- и S-образную линеарную форму. Иногда эта форма ихтиоза у мужчин сочетается с умственной отсталостью (имбецильность) и эпилепсией. Ногтевые пластины могут быть утолщены вплоть до онихогрифоза.
При гистологическом исследовании выявляют гиперкератоз, гранулез, дезорганизацию зернистого слоя и специфическую вакуолизацию клеток зернистого и шиповатого слоев вследствие внутри- и межклеточного отека, акантоз, папилломатоз.
Дифференциальная диагностика. Заболевание необходимо дифференцировать от веррукозного невуса, который, как правило, имеет меньшую распространенность. В сомнительных случаях проводят гистологическое исследование: гранулез и специфическая вакуолизация клеток зернистого и шиповатого слоев характерны для иглистого ихтиоза и отсутствуют при невусе. Синдром Нетертона (Netherton syndromus) —наследственный симптомокомплекс, включающий поражение кожи (ихтиоз, нейродермит) и волос (перекрученные волосы, трихорексис инвагинирующий и др.). Впервые сочетание ихтиоза — ихтиозиформной врожденной эритродермии с поражением волос по типу узловатого трихорексиса описал Е. W. Netherton у больной бронхиальной астмой (1958). Дальнейшие наблюдения показали, что при синдроме Нетертона может возникнуть другая форма ихтиоза — линеарный огибающий ихтиоз, а структурные аномалии волос могут быть разных типов: перекрученные волосы, инвагинирующий трихорексис — поперечные разрывы волос с вколачиванием проксимального отрезка в дистальный, узловатый трихорексис — структурная деформация волос с образованием узлов по типу утолщений на бамбуковых стержнях. Различен и характер атопии при синдроме Нетертона: бронхиальная астма, ангионевротический отек, диффузный нейродермит (атонический дерматит).

При гистологическом исследовании выявляют паракератоз, акантоз со спонгиозом, периваскулярные инфильтраты, состоящие из лимфоцитов, гистиоцитов и полиморфноядерных клеток.
При электронной микроскопии обнаруживают наиболее выраженные изменения в зернистом слое, где выявляют малые гранулы, не связанные с тонофиламентами, и большие, сохраняющие связь с тонофиламентами. Оба типа гранул окружены рибосомами. Тонофиламенты зернистого слоя короткие, расположены в виде скоплений.
Дифференциальную диагностику проводят с ламеллярным ихтиозом, линеарным огибающим ихтиозом, диффузным нейродермитом, а также с изолированными аномалиями развития волос, при которых не наблюдается такой комплексной патологии.
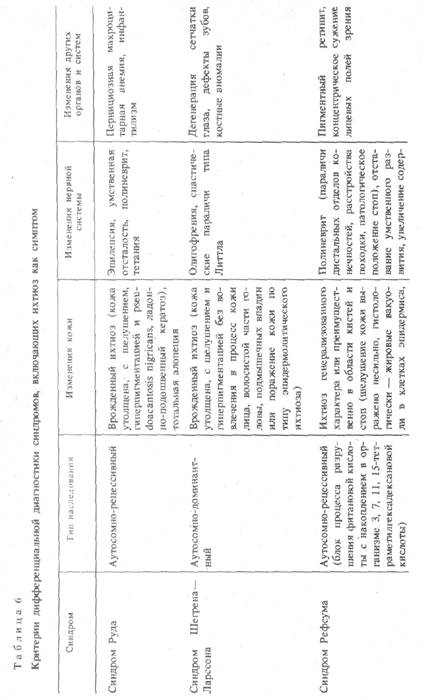
Симптомокомплексы, при которых изменения кожи по типу ихтиоза сочетаются с поражением нервной системы, нередко называют нейроихтиозом. К ним относят синдром Руда, синдром
Шегрена — Ларссона, синдром Рефсума, синдром Попова. Критерии дифференциальной диагностики этих синдромов представлены в табл. 6.
Синдром Руда (syndromus Rud) — наследуемый по аутосомно-рецессивному типу симптомокомплекс — проявление нейроэндо-эктодермальной дисплазии, включающей изменения кожи (врожденный ихтиоз), поражения нервной (эпилепсия, умственная отсталость, полиневрит), а также (не всегда) кроветворной (макроцитарная анемия) и эндокринной (инфантилизм) систем. Описан Е. Rud (1927) под названием «сочетание инфантилизма, тетании, эпилепсии, врожденного ихтиоза, полиневрита, пернициозной анемии». В дальнейшем к синдрому был добавлен еще один симптом — олигофрения. Предполагают, что в основе развития синдрома Руда лежат гипофизарные расстройства. Кожа утолщенная, гиперпигментированная, шероховатая, с явлениями pseudoacantosis nigricaps в складках и ладонно-подошвенным кератозом. Возможна тотальная алопеция.
При гистологическом исследовании выявляют ортокератотический гиперкератоз, гранулез, неравномерный акантоз и папилломатоз.
Этот синдром необходимо дифференцировать от других невро-ихтиозов, в частности аутосомно-рецессивного синдрома Шегрена - Ларссона.
Синдром Шегрена — Ларссона (syndromus Sjogren — Larsson) (син.: синдром Грейтера). Помимо кожной симптоматики, отмечающейся при синдроме Руда, — врожденного ихтиоза (однако при этом кожа лица, волосистой части головы, промежности, подмышечных впадин не поражена) и олигофрении, наблюдается спастическая диплегия типа Литтла, особенно выраженная на верхних конечностях. Возможны дегенеративные изменения сетчатки глаз с резким снижением зрения, дефекты зубов, костные аномалии. Возможно также поражение кожи по типу эпидер-молитического ихтиоза — буллезный ихтиозоформной эритродермии [Desmons E. et al., 1971]. При этом волосы, ногти, слизистые оболочки не поражены. Заболевание наследуется аутосомно-доминантно (плейотропный ген). У родственников больного наблюдаются только неврологические дистрофические дефекты.
Синдром Рефсума (syndromus Refsum) — симптомокомплекс, наследуемый по аутосомно-рецессивному типу, в основе которого лежит нарушение липоидного обмена — блокирование процесса разрушения фитановой кислоты и накопление в организме (сыворотка, ткани) 3, 7, 11, 15-тетраметилгексадексановой кислоты. Синдром Рефсума включает ихтиоз, пигментный ретинит, полиневрит, отставание умственного развития. Наибольшее значение имеют признаки поражения глаз в виде пигментного ретинита (Симптом «перца и соли» на глазном дне с «выцвечиванием» дисков), концентрического сужения лицевых полей зрения. Симптомами полиневрита являются парезы и параличи дистальных отделов конечностей, расстройства походки, патологическое положение стоп. В цереброспинальной жидкости увеличено содержание γ и β-глобулинов. Врожденный ихтиоз характеризуется сухостью и шелушением кожи генерализованного характера или преимущественно в области кистей и стоп. Могут наблюдаться мозжечковые симптомы (атаксия, нистагм) и др. В сыворотке крови больных обнаруживают 3, 7, 11, 15-тетраметилгексадексановую кислоту, обычно отсутствующую в норме, увеличение содержания трансаминаз, церулоплазмина, в моче — увеличение содержания гликозаминогликанов и жирных кислот.
При гистологическом исследовании выявляют фиброзное утолщение мягкой мозговой оболочки с инфильтрацией ее жировыми макрофагами, жировую дегенерацию периферических нервов, дегенеративные изменения клеток передних рогов спинного мозга, в коже, помимо гиперкератоза, отмечаются жировые вакуоли в клетках эпидермиса, окруженные митохондриями.
Синдром Попова (syndromus Popoff). Тип наследования неизвестен. Синдром включает врожденный ихтиоз, карликовый рост, слабоумие, множественный остеопороз.
Врожденная кальцифицирующая хондродисплазия Конради — Хюнерманна (chondrodysplasia calcificans congenita Conradi —Hunermann) наследуется аутосомно-рецессивно. Для синдрома характерны множественные очаги кальцификации в хрящах, низкий рост, короткие конечности, тугоподвижность суставов, седловидный нос, врожденная катаракта, пороки развития сердечнососудистой системы. Кожа туловища и конечностей сухая, с очагами эритемы, гиперкератозом, пятнистой гиперпигментацией, диффузной кератодермией ладоней; выражен гипотрихоз, возможна алопеция. При гистологическом исследовании выявляют выраженный гиперкератоз, глубокие инвагинации расширенных устьев сальных желез.
С возрастом поражение кожи нередко исчезает. При дифференциальной диагностике с различными формами ихтиоза, а также эритрокератодермией врожденную кальцифицирующую хондродисплазию можно отличить по основному признаку заболевания — типичному поражению костной системы, нехарактерному ни для какой другой формы ихтиоза или эритрокератодермии.
Синдром мальабсорбции — симптомокомплекс, развивающийся при нарушении процессов всасывания в желудочно-кишечном тракте. Первичный (врожденный) синдром нарушения всасывания обусловлен генетическим дефектом — отсутствием в пищеварительном тракте ряда ферментов, сопровождающимся изменением слизистой оболочки тонкой кишки (уменьшены толщина и складчатость слизистой оболочки). На коже постепенно развиваются ихтиозиформные изменения — сухость, чешуйки, сопровождающиеся гиперпигментацией кожи (или депигментацией), дистрофическими изменениями ногтевых пластин (продольная или поперечная исчерченность, истыканность) и волос (с развитием диффузной или очаговой алопеции). Характерны также сухость конъюнктивы, глоссит, афтозно-язвенный стоматит.
В ряде случаев синдром мальабсорбции может развиться вторично на фоне необычной кишечной флоры, может сопровождаться, кроме перечисленной симптоматики, телеангиэктатической пурпурой, склероатрофией, ахлоргидрией, нарушением экскреции витамина В12 и всасывания ксилозы [Duperret J., 1974].
При дифференциальной диагностике с вульгарным ихтиозом необходимо иметь в виду поражения слизистых оболочек (конъюнктивит, стоматит, глоссит), не наблюдающиеся при ихтиозе, а также характер патологии тонкой кишки.
Дата добавления: 2017-02-13; просмотров: 4638;
Поиск по сайту

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
