Сера в металле и его десульфурация
Растворимость серы в жидком железе практически не ограничена. В расплаве сера образует с железом ионные группировки S2- и Fe2+, близкие по химическому составу к сульфиду железа FeS. Возможно, что не вся сера в железном расплаве ионизирована и часть ее находится в атомарном (несвязанном) состоянии.
Размеры (радиусы) атомов серы ( 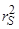 =0,105 нм), а тем более ее ионов (
=0,105 нм), а тем более ее ионов ( 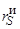 =0,174 нм) слишком велики для образования твердых растворов внедрения в железе, поэтому при кристаллизации сплавов железа происходит резкое сокращение растворимости серы, продолжающееся и при дальнейшем понижении температуры и в особенности при аллотропическом превращении и d–g (рис. 22). Как известно, именно это скачкообразное изменение растворимости серы является основной причиной возникновения пороков в сталях, содержащих [S]>0,04¸0,05 % при их горячей пластической обработке.
=0,174 нм) слишком велики для образования твердых растворов внедрения в железе, поэтому при кристаллизации сплавов железа происходит резкое сокращение растворимости серы, продолжающееся и при дальнейшем понижении температуры и в особенности при аллотропическом превращении и d–g (рис. 22). Как известно, именно это скачкообразное изменение растворимости серы является основной причиной возникновения пороков в сталях, содержащих [S]>0,04¸0,05 % при их горячей пластической обработке.

Растворимость серы в расплавленном железе изменяется в зависимости от концентрации других его примесей. Об этом наиболее удобно судить по изменению активности серы и по соответствующему изменению параметра взаимодействия серы с этими примесными элементами. Увеличение коэффициента активности серы gS и параметра взаимодействия первого порядка того или иного элемента и серы.
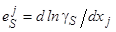
означают уменьшение растворимости серы при данных условиях и, наоборот, уменьшение gS и отрицательное значение 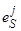 означают удаление раствора серы от насыщения ей металла и рост растворимости серы.
означают удаление раствора серы от насыщения ей металла и рост растворимости серы.
Из диаграммы (рис. 23) и сводки значений параметров взаимодействия серы и других элементов, растворенных в расплавленном железе (при I600 °C) [32], видно, что только углерод и кремний значительно уменьшают растворимость серы в расплавах на основе железа.
Таблица 4 – Значения параметров взаимодействия первого порядка для растворов серы и некоторых других компонентов расплавов на основе железа при 1600 °С [32].
| Элемент | Al | C | Cr | Ce | Mn | O | P | Si | Ti | V | Zr |
|
| 0,035 | 0,11 | -0,011 | 0,914 | -0,02 | -0,27 | -0,29 | 0,064 | -0,072 | -0,016 | -0,052 |
При понижении температуры жидких расплавов на основе железа происходит некоторое понижение растворимости серы, хотя влияние концентрации самой серы на ее активность при понижении температуры несколько уменьшается:
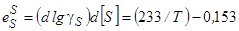 (96)
(96)
В целом это изменение весьма незначительно по сравнений со скачком растворимости серы, наблюдаемым при кристаллизации сплава и превращениях Feж→Feδ и Feδ→Feγ.
 При дальнейшем понижении температуры в областях Feγ и Feα. происходит дальнейшее существенное уменьшение растворимости серы в железе, что является причиной выделения ее в отдельную фазу, т.е. образования "послекристаллизационных" или "четвертичных", иногда ультрамикроскопических включений FeS.
При дальнейшем понижении температуры в областях Feγ и Feα. происходит дальнейшее существенное уменьшение растворимости серы в железе, что является причиной выделения ее в отдельную фазу, т.е. образования "послекристаллизационных" или "четвертичных", иногда ультрамикроскопических включений FeS.
Понижение значений gS и отрицательные значения параметров взаимодействия первого порядка для металлов связаны с образованием или более прочных, по сравнению с FeS, сульфидов. Термодинамические данные об образовании сульфидов некоторых металлов, имеющие значение для производства стали приведены в таблице 5.
Сера является одним из наиболее поверхностно-активных элементов в расплавленном железе, а также в чугунах и практически во всех расплавленных сталях. Как уже отмечалось, максимум величины адсорбции серы в жидком железе и во многих расплавах на его основе колеблется в зависимости от температуры и состава расплава в пределах объемного содержания серы в металле 0,04¸0,07 %.
Таблица 5 – Стандартные изменения энтальпии и энтропии при образовании некоторых сульфидов
| Параметры | Температура, °К | H°, кДж/моль | S°, кДж/моль, °К |
| Feγ+1/2S2газ=FeSж | 1500-1655 | -113,45 | -0,026 |
| Feδ+1/2S2газ=FeSж | 1655-1809 | -111,77 | -0,025 |
| Feж+1/2S2газ=FeSж | 1809-2000 | -125,46 | -0,033 |
| Caж+1/2S2газ=CaSт | 1500-1765 | -525,6 | -0,108 |
| Caгаз+1/2S2газ=CaSт | 1765-200 | -702,9 | -0,193 |
| Mgгаз+1/2S2газ=MgSт | 1500-2000 | -554,7 | -0,198 |
| Mnж+1/2S2газ=MnSж | 1516-1803 | -289,7 | -0,080 |
| 2Alж+3/2S2газ=Al2S3т | 1500-2000 | -688,2 | -0,325 |
| Mnж+1/2S2газ=MnSж | 1803-2000 | -264,1 | -0,066 |
По расчетам С.И. Попеля [57], концентрация серы в моноатомном поверхностном слое в этом случае приближается к ее концентрации в сульфиде железа, т.е. превосходит объемную концентрацию серы почти на три порядка. Именно этим объясняют переход некоторой доли серы в результате ее окисления до SO2 при диспергировании металла в реакционной зоне кислородных конвертеров, т.е. в условиях весьма существенного избытка кислорода.
Этим примером практически исчерпывается экстрагирование серы в газовую фазу, т.к. ее окисление в компактном объеме металла и испарение серы имеют совершенно ничтожное значение. Например, парциальное давление паров серы в равновесии с ее раствором в расплавленном железе ничтожно, его можно определить уравнением:
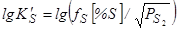 =3760/Т+0,535 (97)
=3760/Т+0,535 (97)
т.е. при 1800 °К 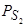 =231 Па.
=231 Па.
Напротив, поглощение серы из газовой фазы плавящимся или уже расплавленным металлом термодинамически весьма возможно и часто имеет место, например, при мартеновском способе производства стали в случае использования для отопления печи виды топлива, содержащие серу – в частности сернистые мазуты, коксодоменные газовые смеси и т.п. [45, 82, 83].
В процессе очищения металла от серы ведущую роль во всех современных металлургических агрегатах, несомненно, играют шлаки. В зависимости от их химического состава и физических свойств удаление серы из металла шлаком может иметь различную природу.
Экстрагирование серы из металла в шлак в той же форме ее существования, в которой она находится в металле, без каких-либо изменений этой формы. В полном смысле слова, такое экстрагирование; представляет собой переход серы в соответствии с "законом распределения" и выравнивания химических потенциалов серы в металле и шлаке. Это имеет место, например, при мартеновской или электроплавке на кислом поду [101, 102]. Как отмечалось ранее, в металле содержащем менее 0,20-0,50% марганца, сера находится в основном в виде FeS и в форме бедных MnS расплавов FeS-MnS. В этом виде она и переходит в шлак.
Однако, растворимость серы в кислом железистом шлаке очень невелика, десятые и сотые доли процента [102]. Поэтому при правке в кисловых агрегатах процесс [FeS]®(FeS) практического значения не имеет. Но при наличии в металле марганца и замены в шлаке некоторой части FеO на MnO ситуация несколько изменяется: в металле группировки FeS заменяются более сложными сульфидными расплавами (FeS)(MnS), температура плавления которых повышается по мере увеличения в них содержания MnS и в этом же направлении уменьшается их растворимость в металле (рис. 24). В то же время повышение (МпО) в шлаках, очевидно, несколько повышает растворимость в них серы. Поэтому наблюдаемый, неравновесный коэффициент распределения серы при плавках в кислых агрегатах и при составах шлаков близких к насыщению их кремнеземом, (S) /[S]=hS колеблется в пределах 0,8-25 [103]. Учитывая поддающуюся оценке и очень зависящую от температуры и соотношения (FeS)/(MnS) растворимость в жидком металле группировок Fe-Mn-S можно считать, что и в присутствии марганца имеет место переход серы из металла в шлак без изменения формы ее существования.
Ранее, когда дефицит марганца в СССP не ощущался так остро, его широко применяли в сталеплавильном производстве в качестве десульфуратора.
Переход серы из металла в практически нерастворимые в нем, но хорошо растворимые в шлаке сульфиды и извлечение последних в шлак.

 К числу элементов, образующих такие сульфиды, относятся прежде всего щелочноземельные металлы – Са, Mg, Ba, Sr. Сложность использования щелочноземельных металлов в качестве десульфураторов связана с их весьма высокой испаряемостью.
К числу элементов, образующих такие сульфиды, относятся прежде всего щелочноземельные металлы – Са, Mg, Ba, Sr. Сложность использования щелочноземельных металлов в качестве десульфураторов связана с их весьма высокой испаряемостью.
Например, упругость пара магния уже при 600 °С составляет 170Па и 0,018 мПа при 1000 °С [80].
Поэтому растворимость щелочноземельных элементов в жидком железе и в расплавах на его основе весьма ограничена. Например, по данным [105] при давлении паров щелочноземельных металлов около 10 Па их растворимость характеризуется данными, приведенными в таблице 6.
Таблица 6 – Характеристика растворимости щелочноземельных элементов
| Элемент | Парциальная молярная теплота растворения, кДж/моль | Растворимость при 1600 °С (в % по массе) |
| Mg | 0,8-1,2 | |
| Ca | 0,040-0,056 | |
| Sr | 0,003 | |
| Ba | 0,005 |
В этой работе было такие установлено существенное влияние отдельных компонентов сплавов железа на растворимость в них щелочноземельных металлов (см. табл. 7), где приведены значения параметров взаимодействия этих элементов с кальцием и магнием.
Таблица 7 – Параметры взаимодействия элементов
с кальцием и магнием
| Элемент | 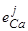
|
| 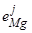
|
|
| Ni | 0,017 | -7,25-10,7 | -6,2 | -0,019 |
| Si | -0,049 | -8,0-10,7 | +0,06 | +0,004 |
| Al | -0,029 | -7,5 | -1,39 | +0,003 |
| C | -0,206 | -1,39 | +0,0009 | |
| Cr | – | – | +0,178 | +0,004 |
Отрицательные значения параметров взаимодействия первого порядка 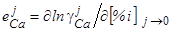 характеризуют понижение активности кальция – например, в присутствии углерода или кремния и, соответственно, увеличения его растворимости в расплаве.
характеризуют понижение активности кальция – например, в присутствии углерода или кремния и, соответственно, увеличения его растворимости в расплаве.
По целому ряду обстоятельств, применение щелочноземельных элементов как десульфураторов передельных чугунов значительно эффективнее, чем использование их в качестве десульфураторов сталей:
а) Прежде всего активность серы в чугунах значительно повышена по сравнению с ее активностью в сталях при постоянной ее концентрации. Это обусловлено высокой концентрацией углерода в чугунах и несколько повышенной концентрацией в них кремния (рис. 29).
б) Упругость паров щелочноземельных элементов над чугунами и соответственно скорость их испарения из чугунов значительно ниже вследствие пониженной примерно на 250-300 °С по сравнению с расплавленной сталью температурой десульфурируемых передельных чугунов. Это означает, что время пребывания щелочноземельных элементов в растворе в несвязанном, свободном состоянии несколько больше и, при ограниченных скоростях массопереноса серы, оно определяет более полное протекание реакций [Mg]+[S]=(MgS) или [Ca]+[S]=(CaS).
в) Величина изменения изобарно-изотермического потенциала окисления всех щелочноземельных металлов, находящихся в элементарном состоянии, значительно более отрицательна, чем при образовании ими сульфидов [61]. В связи с этим как на термодинамическую возможность, так и на кинетику процесса десульфурации положительное влияние оказывает весьма низкая активность кислорода в чугунах – обычно 0,003-0,006. При столь низких активностях кислорода значения DG для обоих сравниваемых процессов сближаются.
г) Время пребывания Са или Mg в растворе в чугуне весьма ограничено. Относительно мало также и время пребывания в нем практически нерастворимых в чугунах сульфидов MgS или CaS. Поэтому значительная доля этих сульфидов выносится на поверхность контакта чугун-шлак за счет конвективных потоков чугуна находясь еще в молекулярно-дисперсном состоянии и переходит в шлак благодаря их высокой растворимости в последнем: kS=(S)/[S]. Часть образующихся сульфидов, зависящая от условий процесса десульфураци, успевает в процессе своего всплывания образовать соответствующие включения, которые иногда наблюдаются при металлографических исследованиях чугунов. При выдержке их в контакте с поглощающими серу шлаками такие включения также быстро ассимилируются шлаком.
Для успешной десульфурации чугунов основная задача металлургов состоит в обеспечении по возможности большей скорости ассимиляции вводимых щелочноземельных элементов металлическим расплавом и, таким образом, опережение их потери в газовую фазу. Это достигается при увеличении поверхности контакта чугуна и вводимых щелочноземельных элементов путем применения, например, магниевой стружки, порошкообразного магния, "магкон".
В США широко применяют карбид кальция, силицид кальция и цианамид кальция, производимых таким образом, что они защищены от гидратации при хранении, но распадаются на мелкие куски при вводе их в контакт с жидким чугуном.
В последние десятилетия в металлургии стали применяются значительно менее, чем кальций и магний распространенные в природе, но также весьма активные по отношению к сере, такие щелочноземельные элементы как барий и стронций и некоторые редкоземельные металлы – церий, ланктан, неодим, празеодим, иттрий. Сульфиды всех этих элементов значительно прочнее, чем сульфид железа, и практически нерастворимы в жидком металле. Однако рассмотрение десульфурирующего влияния этих элементов целесообразно привести в главе, посвященной "облагораживающей" обработке стали перед ее разливкой или даже в процессе кристаллизации, т.е. процессам уже не связанным с экстрагирующим влиянием шлака.
Химические взаимодействия шлаков и металла и серо-поглотительная способность шлаков.

 Практика производства стали в основных сталеплавильных агрегатах показывает постепенное понижение концентрации серы в металле и некоторое повышение ее концентрации в шлаке – даже в те периоды плавки, когда нельзя ожидать образования нерастворимых в металле сульфидов за счет взаимодействия атомов растворенных в жидкой ванне металлов и серы.
Практика производства стали в основных сталеплавильных агрегатах показывает постепенное понижение концентрации серы в металле и некоторое повышение ее концентрации в шлаке – даже в те периоды плавки, когда нельзя ожидать образования нерастворимых в металле сульфидов за счет взаимодействия атомов растворенных в жидкой ванне металлов и серы.
В период использования молекулярной гипотезы строения шлаков это объясняли за счет протекания реакций:
[FeS]+(CaO)=(CaS)+(FeO) или [FeS]+(MnO)=(MnS)+(FeO) или [FeS]+(MgO)=(MgS)+(FeO).
Эти реакции изучались многими исследователями. При этом было твердо установлено, что роль шлаков как десульфурирующей фазы значительно возрастает:
 а) По мере увеличения основности шлака, выражаемой в той или иной форме, с учетом концентраций основных оксидов (в первую очередь СаО) и за счет снижения содержания кислотных оксидов (в основном SiO2).
а) По мере увеличения основности шлака, выражаемой в той или иной форме, с учетом концентраций основных оксидов (в первую очередь СаО) и за счет снижения содержания кислотных оксидов (в основном SiO2).
б)Даже наиболее вероятная из перечисленных выше реакций реакция [FeS]+(CaO)= (CaS) + (FeO) не описывает полностью процесс перехода серы в шлак [55], он более сложен. Поэтому считалось более надежным изучать влияние тех или иных факторов не на константу равновесия этой реакции или двух других, а на условный коэффициент распределения серы между шлаком и металлом:
 hS=(S) /[S]. Это оказалось справедливым не только по отношению к производственным условиям, но также по отношению к лабораторным плавкам, проведенным в довольно строгих условиях [99], рис. 25, 26.
hS=(S) /[S]. Это оказалось справедливым не только по отношению к производственным условиям, но также по отношению к лабораторным плавкам, проведенным в довольно строгих условиях [99], рис. 25, 26.
в) В подавляющем большинстве работ была твердо установлена зависимость условного коэффициента распределения серы – hS от содержания оксидов железа в шлаке и концентрации кислорода в металле (рис.27). Последователи "молекулярной теории" природы металлургических шлаков [113] считали, что процессы экстрагирования серы из металла шлаками и раскисления стали
органически взаимосвязаны. Они изображали этот сложный процесс системой уравнений – например, в случае десулъфурации чугунов известково-глиноземистым шлаком: [MeS]Û(MeS); (MeS)+(CaO)=(CaS)+(MeO) и (MeO)+[C]= =[Me]+{CO}. Несомненным достоинством этой схемы является ее пригодность для экспериментальной проверки хода процесса десульфурации – например, по объему выделенной СО или по изменению исходной концентрации углерода в металле, что хорошо видно из рисунков (28, 29, 30). Таким образом, в настоящее время общепризнанна связь процессов десульфурации и раскисления металлов [106, 107, 114].
 Практика промышленного использования сталей показала, что в ряде конкретных условий их службы эксплуатационные свойства металла тем выше, чем ниже концентрация серы в стали. На этом основании создалось направление производства сталей с весьма низким содержанием серы (в особенности при плавке в дуговых печах). Если в обычных условиях содержание серы в "качественных" сталях менее 0,020-0,030 % считалось нормальным (и было даже утверждено ГОСТами СССP), то в настоящее время часто ставится задача производства стали с содержанием серы 0,005-0,006 %, а иногда даже 0,001-0,003 %. В отдельных случаях это оправдано необходимостью производства крупных слитков и, следовательно, учета ликвидации серы, или же условиями непрерывной разливки и связью склонности металла литых заготовок к обра-зованию "горячих" трещин при повышении содержания в нем серы.
Практика промышленного использования сталей показала, что в ряде конкретных условий их службы эксплуатационные свойства металла тем выше, чем ниже концентрация серы в стали. На этом основании создалось направление производства сталей с весьма низким содержанием серы (в особенности при плавке в дуговых печах). Если в обычных условиях содержание серы в "качественных" сталях менее 0,020-0,030 % считалось нормальным (и было даже утверждено ГОСТами СССP), то в настоящее время часто ставится задача производства стали с содержанием серы 0,005-0,006 %, а иногда даже 0,001-0,003 %. В отдельных случаях это оправдано необходимостью производства крупных слитков и, следовательно, учета ликвидации серы, или же условиями непрерывной разливки и связью склонности металла литых заготовок к обра-зованию "горячих" трещин при повышении содержания в нем серы.


В общем, в последние 40-50 лет развитие металлургии стали поставило ряд новых задач перед теорией десульфурации металла. Ионная теория строения шлаков показала, что переход серы из металла в шлак возможен:
   |
1) по схеме Feж+[S]=Fe2++S2-, имеющей основное значение для слабокислых и невысоко основных и маложелезистых шлаков; или
2) по схеме [S]+O2-=S2-+[O], которая показывает и объясняет взаимосвязь между процессами и десулъфурации металла и его раскисления.
Схема 2 имеет особое значение для десульфурации металла высокоосновными шлаками. Константа равновесия этой реакции была вычислена в первом приближении через концентрации реагирующих компонентов [107]:
lgkS¢=lg([S]×(O2-))/([O]×(S2-))=(6500/T)–2,625; (98)
DG°=–124,57+0,050T кДж/моль. (99)
В настоящее время, благодаря работам В.А. Кожеурова, А.Г. Пономаренко и др. имеется возможность расчета активностей ионов в шлаках и некоторого уточнения уравнений (98) и (99).
Еще Чипманом и др. [104, 107, 108] было установлено, что при температурах и парциальных давлениях в газовой фазе 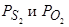 , обычных для плавки в основных мартеновских печах на поверхности контакта газовой фазы и шлака в известной мере возможен процесс:
, обычных для плавки в основных мартеновских печах на поверхности контакта газовой фазы и шлака в известной мере возможен процесс: 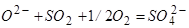 . Более поздние исследования [109,110] показали применимость этого процесса и к условиям электроплавки, в соответствии с чем сера может находиться в шлаке как в сульфидной форме (в виде ионов S6+), так и в сульфатном виде (в форме ионов S2-). Соотношение между активностями этих ионов определяется окислительным потенциалом газовой фазы и, в равновесных условиях, химическим потенциалом кислорода в шлаке (концентрацией оксидов железа в шлаке, его основностью) [81]. Таким образом, при очень низких парциальных давлениях кислорода (
. Более поздние исследования [109,110] показали применимость этого процесса и к условиям электроплавки, в соответствии с чем сера может находиться в шлаке как в сульфидной форме (в виде ионов S6+), так и в сульфатном виде (в форме ионов S2-). Соотношение между активностями этих ионов определяется окислительным потенциалом газовой фазы и, в равновесных условиях, химическим потенциалом кислорода в шлаке (концентрацией оксидов железа в шлаке, его основностью) [81]. Таким образом, при очень низких парциальных давлениях кислорода ( 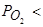 1 Па) распределение серы между газовой фазой и шлаком активно протекает по так называемой сульфидной схеме:
1 Па) распределение серы между газовой фазой и шлаком активно протекает по так называемой сульфидной схеме:
1/2S2газ+(О2-)=О2газ+(S2-), т.е:
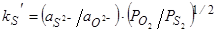 , (100)
, (100)
При более высоких парциальных давлениях кислорода ( 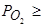 10 Па) распределение серы происходит по сульфатной схеме:
10 Па) распределение серы происходит по сульфатной схеме:
1/2S2газ+3/2O2газ+(O2-)= 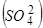 ;
;
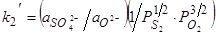 . (101)
. (101)
Сульфидная емкость шлаков была количественно оценена в работе [115], авторы которой для достижения низкого потенциала кислорода в газовой фазе и для контроля в ней потенциала серы применили смеси СО, СО2 и SO2. В работе [116] сульфидная емкость силикатных, преимущественно низкоосновных и кислых шлаков, упрощенно определялась по (см. уравнение 100) уравнению
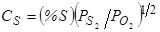 , (102)
, (102)
 что позволило авторам выполнить широкие экспериментальные определения CS при 1550 °С. Они показали, что даже в шлаках системы FeO, MnO, SiO2, близких к насыщению кремнеземом, lgCS составляет 2-2,5 (рис. 31). Сульфидная емкость шлаков имеет существенное значение, например, при основной мартеновской плавке в случае отопления печи серосодержащими видами топлива.
что позволило авторам выполнить широкие экспериментальные определения CS при 1550 °С. Они показали, что даже в шлаках системы FeO, MnO, SiO2, близких к насыщению кремнеземом, lgCS составляет 2-2,5 (рис. 31). Сульфидная емкость шлаков имеет существенное значение, например, при основной мартеновской плавке в случае отопления печи серосодержащими видами топлива.
Сульфатная схема распределения серы между газовой фазой и шлаком имеет гораздо меньшее практическое значение. Поэтому решающую роль в десульфурации металла почти всегда играет переход серы из металла в шлак в форме сульфидов, а следовательно, поглотительная способность СS или емкость шлака по отношению к ионам S2-. В связи с этим определенный интерес представляет процесс 1/2S2газ+(O2-) =1/2O2газ+S2- и характеризующая его равновесие константа k1:
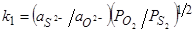 , откуда
, откуда
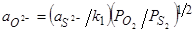 , или
, или
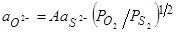 , (103)
, (103)
|
где 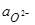 – активность ионов кислорода в шлаке.
– активность ионов кислорода в шлаке.
В соответствии с теорией совершенных ионных растворов характеризует (численно равняясь концентрации свободных ионов О2- за вычетом тех ионов кислорода, которые вошли в состав сложных анионов 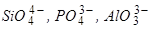 и т.д.) основность шлака. Ее приближенно можно заменить суммой молярных долей основных оксидов(SNосн).
и т.д.) основность шлака. Ее приближенно можно заменить суммой молярных долей основных оксидов(SNосн).
В работе [81] приведен график зависимости величины 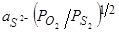 от (при постоянных температурах). Величину
от (при постоянных температурах). Величину 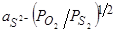 авторы, аналогично [115] и [116] называют сульфидной емкостью шлака, которая характеризует его способность поглощать серу из газовой фазы различного состава (с переменными значениями
авторы, аналогично [115] и [116] называют сульфидной емкостью шлака, которая характеризует его способность поглощать серу из газовой фазы различного состава (с переменными значениями 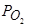 и ). Между сульфидной емкостью шлака и величиной равновесного распределения серы между шлаком и металлом при каждой данной температуре имеется вполне определенная связь.
и ). Между сульфидной емкостью шлака и величиной равновесного распределения серы между шлаком и металлом при каждой данной температуре имеется вполне определенная связь.
Дата добавления: 2016-11-26; просмотров: 3247;
Поиск по сайту
Узнать еще
- A ... метка (без метки) на шатуне (стрелка) для 26.20b Измерение внутреннего диаметра
- Andantino con moto А. Бородин. Для берегов отчизны дальней
- Homo politicus и его роли
- HTML заголовок и его виды
- I-s диаграмма рабочего процесса ГТД
- I.2. Основные категории водопотребления промышленных предприятий и их особенности
- I.5.1 ГРАВИТАЦИОННОЕ ПОЛЕ, ЕГО НАПРЯЖЁННОСТЬ
- I2. Особенности аэродинамики несущего винта (НВ)

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
