ФИЗИКО-ХИМИЧЕСКИЕ И ФИЗИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Любое (физическое, химическое, физико-химическое) явление, в результате которого изменяются свойства системы, может служить основанием для проведения анализа, если есть возможность зафиксировать эти изменения качественно или количественно.
Следует отметить, что многие методы идентификации весьма затруднительно отнести строго к химическим, физико-химическим или физическим. В первом приближении можно считать, что основанием для появления аналитического сигнала в химических методах служит химическая реакция, в физико-химических методах аналитическим сигналом является физическое явление, возникшее в результате химической реакции, а в физических методах фиксируется определённое физическое свойство аналита.
Различают прямые и косвенные методы. В прямых методахданное свойство является критерием содержания вещества, т.е. изучаются соотношения между составом и свойствами. В косвенныхметодах анализа изменение аналитического сигнала указывает на окончание реакции и используется для фиксирования этого момента. Как правило, в инструментальных методах анализа применяются сенсоры (датчики), которые дают информацию о составе и свойствах среды, в которой они находятся.
Инструментальные методы идентификации очень многочисленны. Табл. 6.3 представляет классификацию некоторых из них.
Таблица 6.3
Классификации некоторых физических и физико-химических методов идентификации
| Групповое название методов | Основание для появления аналитического сигнала | Аналитический сигнал, методы идентификации |
| Оптические | Появление спектра в результате взаимодействия электромагнитного излучения с атомами или молекулами анализируемого вещества | Спектры поглощения: · атомно-абсорбционная спектроскопия (спектрофотометрия ) · фотоколориметрия |
| Спектры излучения: · эмиссионная спектроскопия · люминесцентная спектроскопия | ||
| Спектры отражения: · нефелометрия взвесей | ||
| Электрохимические | Окислительно-восстановительные | Электродный потенциал, ЭДС · потенциометрия |
| Электропроводимость · кондуктометрия | ||
| Количество электричества · кулонометрия | ||
| Хроматографические | Сорбция | · сорбционная хроматография |
| Растворимость | · распределительная хроматография | |
| Ионный обмен | · ионообменная хроматография | |
| Термические | Энергетические эффекты физико-химических превращений | Изменение энтальпии · калориметрия |
| Разность температур · дифференциально-термический (ДТА) | ||
| Изменения массы · термогравиметрический (ТГА) | ||
| Ядерно-химические | Образование радиоактивных изотопов | · радиоактивационный |
Спектральные методы количественного анализа
Спектроскопия базируется на квантовой теории, согласно которой атому или молекуле в стационарном состоянии соответствует некоторая последовательность энергетических уровней. Переход частицы из одного состояния в другое сопровождается испусканием или поглощением кванта электромагнитного излучения, которое регистрируется соответствующими приборами в виде спектра.
В оптических спектроскопических методах исследования изучаются спектры электромагнитного излучения длиной волны 10-8…10-3 м, которое испускается, поглощается или рассеивается веществом. Абсорбционный анализ - это анализ по поглощению света в видимой, ультрафиолетовой и инфракрасной областях спектра. На поглощении света в видимой части cпектра истинными растворами основаны фотоколориметрия и спектрофотометрия; взвесями - турбидиметрия, на рассеянии света взвесями - нефелометрия.
Сущность атомно-абсорбционной спектроскопии состоит в том, что при взаимодействии со световой энергией в атомах поглощающего вещества происходит переход валентных электронов на более высокие энергетические уровни. Электронные переходы, вызванные поглощением строго определенных квантов световой энергии, характеризуются наличием строго определенных полос поглощения в спектрах.
Фотометрические методы анализа, к которым относятся фотоколориметрия и спектрофотометрия,применяются для изучения состава и свойств строительных материалов. На спектрографе или спектрофотометре получают спектр, в котором длина волны является качественной характеристикой вещества и зависит от природы.
Прибор для проведения исследования содержит пять основных узлов:
1) источник излучения;
2) монохроматор, где выделяется длина волны, при которой происходит наиболее интенсивное поглощение;
3) исследуемый образец;
4) приёмник излучения;
5) устройство, регистрирующее спектр.
В фотоколориметрии проводится сравнение интенсивности окраски исследуемого и стандартного (с точно известной концентрацией) растворов. Основные узлы фотоэлектроколориметра представлены на рис. 6.3.

Рис. 6.3. Схема основных узлов фотоэлектроколориметра
Основной закон фотометрии (закон Бугера-Ламберта-Бера)устанавливает зависимость уменьшения интенсивности монохроматического светового потока It , прошедшего через слой окрашенного раствора концентрацией с и толщиной l, от интенсивности падающего света I0 .
It = I0 · 10 -ε l C ,
где ε - молярный коэффициент поглощения раствора концентрации 1моль/дм3, зависящий от длины волны падающего света, природы растворенного вещества, температуры.
Преобразуем уравнение, взяв отношение I0 : Itи прологарифмировав обе части уравнения
(I0 : It) = 10 ε l C ; lg ( I0 : It ) = lg ( 10 ε lC )
Отношение (I0 : It) = D называется оптической плотностью. В результате преобразования выражения получим
D= ε lC,
откуда следует, что оптическая плотность прямо пропорциональна концентрации окрашенного вещества и толщине поглощающего слоя.
Сущность электрохимических методов анализа
Электрохимические методы исследования основаны на процессах обмена ионов или перехода электронов, протекающих на поверхности электрода или в приэлектродном пространстве. Аналитическим сигналомслужит любой электрический параметр (потенциал, сила тока, сопротивление и т.д.), величина которого зависит от состава исследуемой системы. В потенциометрическомметоде исследования измеряют электродвижущую силу (ЭДС) обратимых электрохимических систем, для чего необходимо наличие двух электродов: индикаторного и электрода сравнения. В зависимости от алгебраической величины электродных потенциалов величина ЭДС определяется по формулам:
ЭДС = Е индикаторного электрода ─ Е электрода сравнения,
ЭДС = Е электрода сравнения ─ Е индикаторного электрода.
Индикаторный электрод обнаруживает и фиксирует аналитический сигнал, а электрод сравнения помогает измерить этот сигнал.
Потенциал индикаторного электрода зависит от активности исследуемых веществ, потенциал электрода сравнения не меняетсяпри проведении исследований. В качестве последних часто используют хлоридсеребряный, а также каломельный электроды, потенциалы которых при постоянной концентрации Сlˉ остаются неизменными.
Особую группу индикаторных электродов составляют ионоселективные (мембранные) электроды, потенциал которых в зависимости от типа мембраны и может определяться активностью ионов Н+, Са2+, Мg2+, Na+, K+, Cl-, NH4+, NO3-, PO43-, CO32- и т.д. В частности, потенциал стеклянного электрода зависит от активности ионов водорода ан+ :
Естеклянного электрода = К + lg ан+,
где К - постоянная, определяемая сортом стекла и конструкцией электрода.
Поскольку рН = – lg а н+ , то с помощью индикаторного стеклянного электрода можно измерять рН растворов и суспензий.
Кондуктометрические методы основаны на измерении электропроводимости. В области сравнительно невысоких концентраций эквивалентная электрическая проводимость электролитов λ, [ Cм · м2 · моль-1] обычно растет с уменьшением концентрации раствора. На существовании линейной зависимости между электропроводимостью и концентрацией основана прямая кондуктометрия.
В методе кондуктометрического титрованиямогут быть использованы химические реакции, в ходе которых достаточно заметно изменяется электрическая проводимость и в точке эквивалентности изменение происходит скачкообразно.
Кулонометрическийметод идентификации основан на законе М. Фарадея, согласно которому масса, образующегося на электродах вещества (испытавшего электрохимическое превращение) прямо пропорциональна количеству прошедшего электричества:
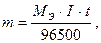
где m ─ масса вещества, окисленного или восстановленного на электроде, г;
М э ─молярная масса эквивалента вещества, г/моль; I ─ сила тока, А;
t ─ время электролиза, с; 96500 ─ число Фарадея, Кл/моль (А · с/моль).
Сущность хроматографических методов анализа
Хроматография(от греч. chromatos − цвет и grapho − пишу) − физико-химический метод разделения и анализа смесей, основанный на распределении их компонентов между двумя фазами — неподвижной (сорбент) и подвижной (элюент), протекающей через неподвижную.
Классификация хроматографических методов представлена на рис. 6.4.

Рис. 6.4. Классификация хроматографических способов разделения
Более подробно хроматографические процессы можно описать также следующим образом.
1. В зависимости от природы взаимодействия, обусловливающего распределение компонентов между элюентом и неподвижной фазой, различают:
∙ адсорбционную (различная сорбируемость разделяемых веществ адсорбентом);
∙ распределительную (разная растворимость компонентов смеси в неподвижной фазе и элюенте);
∙ ионообменную (различные константы ионообменного равновесия между неподвижной фазой (ионитом) и компонентами разделяемой смеси;
∙ молекулярно-ситовую (разная проницаемость молекул компонентов в неподвижную фазу;
∙ осадочную (различная способность разделяемых компонентов выпадать в осадок на твёрдой неподвижной фазе).
2. Различают колоночную и плоскостную хроматографию:
∙ в колоночной сорбентом заполняют специальные трубки — колонки, а подвижная фаза движется внутри колонки благодаря перепаду давления;
∙ плоскостная хроматография подразделяется на тонкослойную и бумажную.
3. В зависимости от способа перемещения разделяемой смеси вдоль слоя сорбента используется:
∙ фронтальная (рис. 6.5, а), когда в слой сорбента непрерывно вводится разделяемая смесь, состоящая из газа-носителя и разделяемых компонентов, например 1, 2, 3, 4, которая сама является подвижной фазой. Через некоторое время после начала процесса наименее сорбируемый компонент (например, 1) опережает остальные и выходит в виде зоны чистого вещества раньше всех, а за ним в порядке сорбируемости последовательно располагаются зоны смесей компонентов: 1 + 2, 1 + 2 + 3, 1 + 2 + 3 + 4;
∙ проявительная (рис. 6.5,б), когда через слой сорбента непрерывно проходит поток элюента и периодически в слой сорбента вводится разделяемая смесь веществ. Через определённое время происходит деление исходной смеси на чистые вещества, располагающиеся отдельными зонами на сорбенте, между которыми находятся зоны элюента;
∙ вытеснительная (рис. 6.5, в), когда в сорбент вводится разделяемая смесь, а затем поток газа-носителя, содержащего вытеснитель (элюент), при движении которого смесь через некоторый период времени разделится на зоны чистых веществ, между которыми окажутся зоны их смеси.
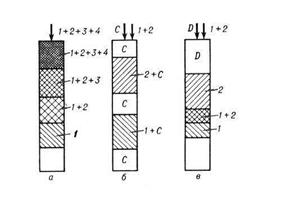
Рис. 6.5. Хроматографический процесс (варианты проведения)
4. по средам, в которых производится разделение, хроматография может быть:
∙ газовая, где в качестве элюента (газа-носителя) используются гелий, азот, аргон и др. газы, а качестве сорбента (частицы диаметром 0,1—0,5 мм) используют силикагели, алюмогели, молекулярные сита;
∙ жидкостная колоночная, в качестве элюента применяют легколетучие растворители (например, углеводороды, эфиры, спирты), а в качестве неподвижной фазы — силикагели.
Хроматография широко применяется в лабораториях и в промышленности
∙ для качественного и количественного анализа многокомпонентных систем;
∙ контроля производства, особенно в связи с автоматизацией многих процессов;
∙ для препаративного (в т. ч. промышленного) выделения индивидуальных веществ (например, благородных металлов);
∙ разделения редких и рассеянных элементов.
Сущность термических методов анализа
Термические методы анализа основаны на взаимодействии вещества с тепловой энергией. Наибольшее применение в аналитической химии находят термические эффекты, которые являются причиной или следствием химических реакций. В меньшей степени применяются методы, основанные на выделении или поглощении теплоты в результате физических процессов. Это процессы, связанные с переходом вещества из одной модификации в другую, с изменением агрегатного состояния и другими изменениями межмолекулярного взаимодействия, например, происходящими при растворении или разбавлении.
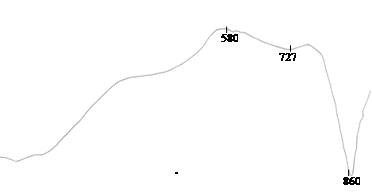
Результаты термических методов анализа представляют в виде термограмм (рис. 6.4.)
 |
| Изменение температуры | |
| Температура |
Рис. 6.6. Дериватограмма зерна-уноса (отхода цементного производства)
Сущность рентгеновских методов анализа
Во многих природных и синтезированных, технически важных материалах кристаллическое вещество находится в виде поликристалла и важно иметь возможность изучить его структуру и свойства именно в таком состоянии. Поликристаллический материал состоит из множества мелких кристалликов. Это может быть или агрегат плотно сцепленных между собой мелких кристаллов (например, металлы, сплавы, многие минералы, керамические материалы), или измельченный порошок данного вещества. Поликристаллическое вещество может состоять из кристалликов различных фаз.
С помощью рентгенографии на поликристаллических образцах можно решать следующие задачи:
- структурный анализ несложных структур;
- определение элементарной ячейки неизвестного вещества;
- исследование фазовых переходов, изучение состояния твердого тела (кристаллическое, аморфное, аморфное с кристаллическими включениями);
- исследование фазового состава вещества (качественный и количественный анализы):
качественный - идентификация кристаллических фаз на основе присущих им значений межплоскостных расстояний dhkl и интенсивности линий Ihkl рентгеновского спектра;
количественный - определение количества тех или иных фаз в смеси:
- определение средних размеров кристаллов, зерен в образце, функции распределения их по размерам, по анализу профиля линий;
- изучение внутренних напряжений: проводят анализ профиля дифракционных линий и сдвига положения этих линий;
- изучение текстур, т.е. характера преимущественной ориентации кристаллитов.
Количественный рентгеновский фазовый анализ основан на зависимости интенсивности дифракционного отражения от содержания Xi соответствующей фазы. Сравнивая экспериментальные значения Ihkl с эталонными и вводя необходимые поправки на поглощение, можно определять содержание фазы Xi . Рентгенограммы имеют вид, представленный на рис. 6.7.
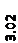 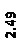 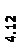 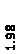 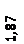     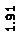 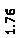 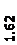 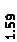       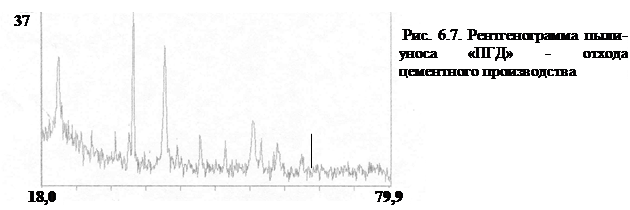 |
Основная литература:
1. Коровин Н.В. Общая химия. М.: Высшая школа, 2008. – 557 с. §§ 16.1– 16.3
Дата добавления: 2016-07-27; просмотров: 3474;
Поиск по сайту
Узнать еще
- I. История открытия и методы исследования вирусов
- II. Категории и методы политологии.
- II. Физические характеристики участников коммуникации
- III и IV нейроны слухового пути. Третьи и четвертые нейроны слухового проводящего пути. Ядра слухового анализатора. Признаки поражения слухового пути.
- III. Методы искусственной физико-химической детоксикации.
- І. Анализаторы І сигнальной системы
- ІІ. Анализаторы ІІ сигнальной системы
- А) Физические свойства минералов

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
