Развития учения о валентности
Валентность (от лат. valentia - сила), - одно из фундаментальных понятий теории химического строения. Оно формировалось вместе с понятием химической связи, параллельно с развитием синтетической химии и методов исследования строения и свойств веществ, и его содержание неоднократно расширялось и изменялось по мере того, как экспериментальная химия находила всё новые и новые классы соединений с неизвестными ранее типами взаимодействия атомов в молекуле, а в последние 30-40 лет - с развитием квантовой химии. В настоящее время накопленный химией экспериментальный материал столь обширен и разнообразен, а картина химической связи в разных соединениях столь пестра, что задача нахождения последовательного, единого и всеобъемлющего определения В. представляется крайне сложной. Эти трудности побуждают некоторых химиков вообще отказаться от поисков универсального понятия В. и заменить его набором более узких, но зато более конкретных и более точных понятий (ковалентность, гетеровалентность, координационное число и т.д.), область применимости каждого из которых ограничена соединениями с каким-либо одним преобладающим типом взаимодействия (ковалентным, ионным, координационным и т.д.). Однако до настоящего времени и в специальной, и в учебной литературе В. продолжает широко использоваться и как определение способности атома к образованию связей в самом общем смысле слова, и как количественная мера этой способности, и как синоним предлагаемых более узких понятий.
Единое и последовательное определение В. следует искать в рамках квантовохимической теории молекулярных орбиталей.
Глава 1. Эволюция понятия "валентность" и его роль в истории химии
В начале 19 в. Дж. Дальтоном был сформулирован закон кратных отношений, из которого следовало, что каждый атом одного элемента может соединяться с одним, двумя, тремя и т.д. атомами другого элемента (как, например, в окислах азота -N2O, NO, N2O3, NO2 и N2O5). В середине 19 в., когда были определены точные относительные веса атомов (И. Я. Берцелиус и др.), стало ясно, что наибольшее число атомов, с которыми может соединяться данный атом, не превышает определённой величины, зависящей от его природы. Например, атом F может соединяться лишь с одним атомом Н, О - с двумя, N - с тремя, С - с четырьмя, образуя соответственно HF, H2O, NH3 и CH4. Два или четыре атома Н в метане CH4 могут быть замещены одним или двумя атомами О с образованием формальдегида CH2O и двуокиси углерода CO2 соответственно, три атома Н в CH4 могут замещаться одним атомом N с образованием цианистого водорода HCN, и т.д. Эта способность связывать или замещать определённое число других атомов и была названа "Валентность" (Э. Франкленд, 1853).
В таком определении В., естественно, всегда выражается целыми числами. Поскольку в то время для водорода не были известны соединения, где он был бы связан более чем с одним атомом любого другого элемента, атом Н был выбран в качестве стандарта, обладающего В., равной 1. В "водородной" шкале кислород и сера имеют В., равную 2, азот и фосфор 3, углерод и кремний 4. Однако "водородной" шкалы оказалось недостаточно: в других соединениях, например в окислах, один и тот же элемент может реализовать В., которые не осуществляются в гидридах (существуют окислы P2O5, SO3 и Cl2O7, но неизвестны гидриды PH5, SH6 и ClH7). В качестве второго стандарта с В., равной 2, был выбран кислород.
В конце 50-х гг. 19 в. А. С. Купер и А. Кекуле постулировали принцип постоянной четырёхвалентности углерода в органических соединениях. Представления о В. составили важную часть теории химического строения А. М. Бутлерова (1861). Образование химической связи рассматривалось как результат взаимного насыщения двух В. пары взаимодействующих атомов (по одной В. от каждого), кратные связи соответствовали насыщению нескольких В. от каждого атома, и т.д. Каждая связь считалась локализованной между двумя атомами и изображалась одной чертой, соединяющей эти атомы. Молекулы стали изображать с помощью структурных формул, получивших особенно широкое распространение в органической химии.
Положения Бутлерова в дальнейшем легли в основу структурной теории, рассматривающей и пространственное расположение атомов в молекуле. Было найдено, что простые молекулы типа MXk с одинаковым центральным атомом M и разными заместителями Х имеют схожее геометрическое строение. Независимость геометрического строения от типа связи в широких пределах привела к мысли, что пространственное расположение атомов в молекулах MXk определяется В. центрального атома М и что эти В. имеют направленный характер.
Периодический закон Д. И. Менделеева (1869) вскрыл зависимость В. элемента от его положения в периодической системе. Элементы одинаковых групп системы обладают одинаковой высшей В., в большинстве случаев равной номеру той группы, в которой находится этот элемент; высшая В. меняется на 1 при переходе от одной группы к соседним. Эта зависимость сыграла чрезвычайно важную роль в развитии химии: зная лишь положение элемента (в том числе элементов, которые в то время ещё не были открыты) в периодической системе, можно было определить его валентные возможности, предсказать состав его соединений и впоследствии синтезировать их. С помощью представлений о формальной (стехиометрической) В. химикам удалось обобщить и систематизировать огромный экспериментальный материал по строению, стехиометрическому составу и свойствам многих десятков и сотен тысяч органических и неорганических соединений.
Первые электронные теории ковалентности и гетеровалентности. До электронных представлений о строении вещества В. трактовалась формально. Лишь в 20 в. было установлено, что химическая связь осуществляется за счёт электронов внешних (валентных) оболочек атомов.
В 1916 Г. Льюис постулировал, что химическая связь осуществляется парой электронов, принадлежащих одновременно обоим взаимодействующим атомам. В 1917 В. Коссель выдвинул гипотезу, согласно которой электронная пара связи переходит целиком к одному из атомов с образованием ионной пары катион - анион, удерживающихся в молекуле электростатическими силами. Согласно обеим гипотезам наиболее устойчивыми оказываются соединения, в которых валентные электроны распределялись так, чтобы каждый атом был окружен оболочкой, имитирующей электронную оболочку ближайшего инертного газа (правило октета). Гипотеза Льюиса положила начало электронной теории ковалентной связи и ковалентности, гипотеза Косселя - теории ионной связи и гетеровалентности. Обе представляли крайние случаи общей картины полярной связи, когда электронная пара смещена к одному из атомов лишь частично и степень смещения может варьировать от 0 до 1. В. атома в соединении, согласно классической электронной теории, равна числу его неспаренных электронов, участвующих в связях, а максимальная В. - обычно полному числу электронов в его валентной оболочке, то есть номеру труппы периодической системы, в которой находится элемент. Элементы одинаковых групп имеют одинаковое число валентных электронов, а внутри одинаковых подгрупп - и одинаковые или очень близкие электронные конфигурации. Сходство строения валентных оболочек атомов обусловливает сходство их соединений.
Ковалентность и гетеровалентность отражают специфику соответствующего типа химической связи. Для ковалентности важна насыщаемость связей, обусловливающая существование молекул в виде дискретных частиц с определённым составом и структурой. Ковалентность эффективна для органических и большинства простых неорганических соединений. Напротив, в случае гетеровалентности максимальное число ионов противоположного знака, способное разместиться вокруг данного иона, в основном определяется соотношениями их размеров. Ионная В. эффективна для сравнительно ограниченного класса соединений, в основном для различных солей щелочных, щёлочноземельных и некоторых др. металлов.
В. в комплексных соединениях. Ещё в конце прошлого века было найдено (А. Вернер, 1893), что многие соединения, как с максимальными (насыщенновалентные), так и с промежуточными В., типа ВСl3, SiCl4, PCl5, CrCl3 и т.п., обладают склонностью к взаимодействию с другими насыщенновалентными соединениями - солями, окислами, молекулами типа H2O, NH3 и др., с образованием довольно прочных комплексных соединений - K[BCl4], K2[SiCl6], NH4[PCl6] и т.д. Исследования их строения рентгеновскими методами показали, что в комплексных анионах и катионах атомы лигандов Х обычно находятся в вершинах правильных многоугольников (октаэдра, тетраэдра и др.), а все связи М - Х одинаковы.
Для представлений о В. комплексные соединения необычны тем, что в них координационное число КЧ может быть больше общего числа валентных электронов атома М. Более того, в парамагнитных комплексах переходных и редкоземельных металлов - K4[CrF6], K3[CrF6], K2[CrF6] и др., некоторые электроны валентной оболочки остаются неспаренными и локализованными у центрального атома и практически не участвуют в связи. Классическая В. и КЧ, как правило, не совпадают, а способность к образованию октаэдрических и тетраэдрических, комплексов оказалась чрезвычайно распространённой и типичной для многих металлов и неметаллов, связанной сложной зависимостью с положением элемента в периодической системе и его В. в исходном простом соединении.
Поэтому было высказано предположение, что, наряду с "классической" В., которая реализуется в исходных простых соединениях типа ВСl3, SiCl4 и др., атомы обладают также "координационной" В., которая насыщается в комплексных соединениях. Предпринимались попытки описать связь в комплексных соединениях в рамках ионной теории, в которой считается, что анионы типа [PF6]- и [МnO4]- построены из ионов P5+ + 6F- и Mn7+ + 4O2- и что В. центрального атома совпадает с зарядом его иона. Однако затраты энергии, необходимой для перевода 1 атома Mn и 4 атомов О в состояния Mn7+ и O2-, далеко не компенсируются выигрышем в энергии при образовании связи. С появлением экспериментальных методов определения эффективных зарядов стало ясно, что эффективные заряды вообще редко превышают значения +1 или +2 у положительно заряженных и -1 у отрицательно заряженных атомов и обычно выражаются дробными долями заряда электрона (в перманганатном анионе заряд на Mn составляет лишь +1,5-+2,0 электрона). Поэтому ионная теория для большинства неорганических соединений, простых и комплексных, не может считаться корректной.
Успехи химии 20 в. и проблемы теории В. В 20 в. экспериментальной химией было синтезировано и изучено строение множества новых соединений, которые также оказалось невозможно уместить в рамки классических представлений о В. Оказалось, что склонность к образованию координационных соединений и насыщению координационных В. вообще чрезвычайно распространена и характерна практически для всех элементов и что суждения о В. на основании одного лишь стехиометрического состава очень часто оказываются несостоятельными без точных данных о структуре соединения и геометрическом расположении ближайшего окружения рассматриваемого атома. По мере развития структурных методов стало известно, что многие соединения с простым брутто-составом (AlCl3, PdCl2, MoO3 и др.), ранее считавшиеся простыми, в действительности даже в парах имеют димерное и полимерное строение - Al2Cl6, (PdCl2) x (рис. 1, а, б), (MoO3)2-5. В них "мостиковые" лиганды, соединённые одинаковыми связями с двумя атомами металлов (на рис. 1 они помечены цифрой 2), обладают координационным числом КЧ = 2. У соединений в твёрдом состоянии, которые часто построены ещё сложнее, КЧ галогенов и кислорода, ранее выбранного в качестве стандартного двухвалентного элемента, могут быть 3 и даже 4. В бороводородах каждый "мостиковый" атом водорода, считавшегося ранее стандартным одновалентным элементом, связан одинаковыми связями с двумя атомами бора (рис. 1, в). Алкильные группы также способны образовывать мостиковые связи в металлоорганических соединениях типа Al2(CH3)6 (рис. 1, г) и др.
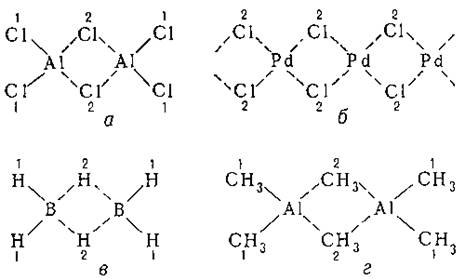
Рис. 1. Мостиковые лиганды (Cl, H, CH3) в димерных и полимерных соединениях: Al2Cl6 (а), (PdCl2)x (б), В2Н6 (в) и Al2(CH3)6 (г)
Для соединений переходных и ряда непереходных элементов оказалось характерным использование дополнительной В. за счёт образования связей металл - металл (кластерные соединения), при этом расстояние между атомами металлов оказалось значительно меньшим, чем в индивидуальных металлах. Например, в дигалогенидах молибдена и вольфрама во многих химических реакциях сохраняется неизменной группа (рис. 2), в которой атомы металла (Me) образуют правильный октаэдр; каждый атом Me связан с четырьмя другими атомами Me и с четырьмя атомами галогена (Hal), а каждый атом Hal связан с тремя атомами Me. Связи Me - Me в кластерах могут быть кратными (как, например, в , где расстояние Re - Re на 0,5 меньше, чем в металлическом Re, и на их образование атомы могут тратить не одну, а несколько В.
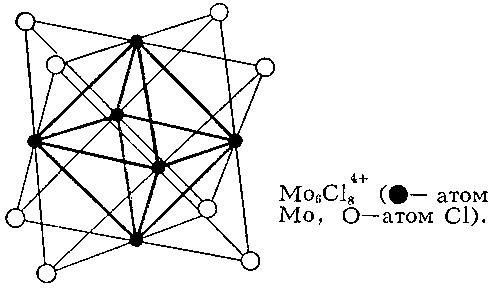
Рис. 2. Кластерная структура Mo6Cl84+
Недостаточность классического понимания В. видна также на примере так называемых "нуль-валентных" соединений, где атом металла связан исключительно с нейтральными молекулами; таковы карбонилы металлов типа Ti(CO)7, Cr(CO)6, Fe(CO)5, аммиакаты типа Pt(NH3)4 и т.д. В них вообще отсутствует классическое валентное взаимодействие (у атомов С и N в молекулах CO и NH3 нет неспаренных электронов), а связь осуществляется только за счёт координационных В. атома металла и молекул лигандов. Нейтральные лиганды часто оказываются мостиковыми и образуют по две, например в Co4(CO)12, и даже по три, например в Rh6(CO)16, связи.
Для теории В. особый интерес представляют так называемые p-комплексы переходных металлов с ароматическими молекулами или молекулами с сопряжёнными связями в качестве лигандов (этиленом, циклопентадиенилом, бензолом и др.) типа ферроцена Fe(C5H5)2, дибензолхрома Cr (C6H6)2 (рис. 3, а, б), тетрациклопентадиенила титана Ti(C5H5)4 и др. В отличие от комплексов типа [Сr (NH3)6]3+, [Сr (H2O)6]2+ или Cr (CO)6, где центральный атом осуществляет связь с лигандом через один атом от каждого лиганда (через N - в аммиакатах, через О - в гидратах, и т.д.), в p-комплексах атомы Fe, Cr и Ti взаимодействуют совершенно одинаково со всеми атомами С каждого ароматического кольца. Непригодность классической В. или КЧ здесь очевидна: при этом пришлось бы считать все атомы углерода 5-валентными, а атомы Fe, Cr и Ti - соответственно 10-, 12- и 20-валентными. Единственный неспаренный электрон, который имеется у радикала ×C5H5 (так же как и у многих других ароматических радикалов типа тропила ×C7H7 и т.д.), в равной степени принадлежит всем углеродным атомам кольца. Для этого класса соединений потребовались представления о делокализованной ("групповой") В., характеризующей всю совокупность атомов С в ароматическом кольце.
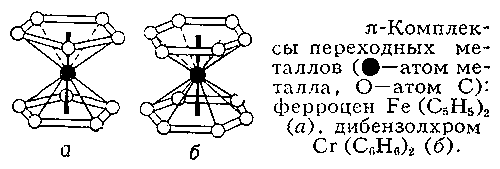
Рис. 3. (π-Комплексы переходных металлов Fe(C5H5)2 (а), дибензолхром Cr(C6H6)2 (б)
Сейчас стало ясно, что КЧ в комплексах, так же как В. в простых соединениях, не является жестко специфической характеристикой элемента: для большого числа металлов были найдены комплексы со всеми промежуточными значениями КЧ от 3 до 7, 8 и 9. При этом возникли трудности с самим определением КЧ: в низкосимметричных высококоординационных комплексах расстояния М - Х, даже для одинаковых лигандов X, часто оказываются неодинаковыми; при этом они могут быть больше тех достаточно коротких расстояний, при которых наличие сильного взаимодействия бесспорно, но всё же недостаточно велики, чтобы их можно было уверенно исключить из координационной сферы комплекса.
Новые проблемы В. возникли и в других разделах химии. Сильное развитие получила химия свободных радикалов (например, метил ×CH3, трифенилметил ×C (C6H5)3 и др.), в которых имеются атомы 3-валентного углерода. В последнем десятилетии были синтезированы соединения инертных газов типа XeF2, XeF4, XeF6, XeO3 и др., то есть соединения элементов, которые ранее считались вообще неспособными к химическому взаимодействию. Стало ясно и то, что В. элементов может сильно меняться с изменением внешних условий, в частности температуры. Например, PCl5, существующий при умеренных температурах в газовой фазе в виде мономерных молекул, при конденсации диспропорционирует, давая пару катион [РСl4]+ (КЧ = 4) - анион [РСl6]- (КЧ = 6). Наоборот, при повышении температуры обнаруживаются молекулы PCl3, PCl2, PCl, ионы PCl4+, PCl3+, Pd2+, PCl+ и т.д. Благодаря успехам химии молекул в газовой фазе за последние 20 лет найдено огромное число соединений (часто сложного состава) с промежуточными и необычными В., которые не обнаруживаются у соединений в обычных условиях. Например, кроме давно известных анионов типа CO32- и SO42-, сейчас обнаружены анионы CO3-, SO4- и нейтральные молекулы CO3, SO4. Кроме насыщенных молекул типа CH4, C2H6, найдены ионы типа CH5+, C2H7+, кроме молекулы H2 - ион Н3+, и т.д.
Сейчас установлено, что подавляющее большинство элементов может проявлять переменную В., образуя весь ряд "валентноненасыщенных" соединений со всеми значениями В. от 1 до максимальной с изменением на 1 (например, известны молекулы BF, BF2 и BF3; CF, CF2, CF3 и CF4 и т.д.). В. не может считаться жестко специфической характеристикой элемента, можно говорить лишь об относительной типичности или относительной устойчивости разных значений В. У непереходных элементов чётных и нечётных групп наиболее устойчивы соответственно чётные и нечётные В., например в молекулах типа PF3, PF5, SF2, SF4, SF6, IF, IF3, IF5, IF7 и т.д., где типичная В. атомов Р, S и I изменяется на 2 единицы.
Радикалы типа ЇPF4, ЇSF3, ЇSF5, ЇIF2, ЇIF4 и т.д. с четырёхвалентным фосфором, нечётновалентными аналогами серы и инертными газами и чётновалентными галогенами значительно менее стабильны, обладают отчётливо выраженной склонностью к отщеплению одного электрона (с образованием более устойчивых катионов типа PF4+, SF3+, SF5+, IF2+, IF4+) или одного атома заместителя и характеризуются значительно меньшими временами существования. У элементов побочных групп соотношения между типичными и менее типичными В. имеют более сложный характер.
Изучение электронных спектров показало, что двухатомные молекулы типа O2, S2, OS и др. имеют два неспаренных электрона; в рамках классических представлений это следовало бы интерпретировать так, будто в подобных молекулах каждый атом сохраняет неиспользованной одну свою В., хотя нет никаких видимых препятствий для их использования.
До сих пор не решена проблема В. в случае интерметаллических соединений, имеющих обычно сложный состав типа Cu5Zn8, Cu31Sn8, Zn21Fe5, нестехиометрических окислов, нитридов, карбидов, силицидов и других соединений металлов, в которых состав может меняться непрерывно в сравнительно широких пределах.
Таким образом, поиск общего определения В., охватывающего все известные типы соединений и тем более способного предсказать возможность или принципиальную невозможность существования ещё не известных классов соединений, представляет сложную проблему. Конечно, параллельно с "неклассическими" соединениями химиками были синтезированы многие сотни тысяч соединений, которые могут быть интерпретированы в рамках обычных классических представлений о В. Однако ясно, что все существующие частные определения В. ограничены определёнными классами и типами соединений, в которых преобладает какой-либо один тип химического взаимодействия. В общем же случае связи имеют промежуточный характер между чисто ионными и чисто ковалентными, в них принимают участие все типы взаимодействия одновременно, но в различных количественных соотношениях, резко изменяющихся от класса к классу и более плавно - от соединения к соединению внутри одного класса. При отсутствии общего определения В. трудность заключается в том, чтобы определить границы, где перестаёт быть справедливым одно частное определение В. и его заменяет другое. Решить эту проблему только на основании экспериментальных фактов и классических представлений невозможно. Существенную помощь здесь может оказать квантовая теория химической связи и В.
Дата добавления: 2016-05-30; просмотров: 4775;
Поиск по сайту
Узнать еще
- Cпособы получения частиц порошков
- Filariidae:. Onchocerca volvulus Систематика, морфология, цикл развития, патогенное действие, диагностика и профилактика онхоцеркоза и лоаоза.
- II. Завоевание Китая маньчжурами. Экономическое положение страны в XVII – начале XIX вв.: аграрная политика Цинской династии, особенности развития городского ремесла
- II. Особенности развития турецкой буржуазии. Становление младотурецкого движения
- II. Политические учения античности
- III Всебелорусское собрание. Программа социально-экономического развития Республики Беларусь на 2006 – 2010 гг.
- III. ТРЕТИЙ ЭТАП ОБУЧЕНИЯ
- IV. Внешние условия развития отрасли информационных технологий

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
