Глава 2. Современные квантово-химические представления о валентности
Начиная с 30-х гг. 20 в. представления о природе и характере В. постоянно расширялись и углублялись, параллельно с расширением и углублением представлений о химической связи. Существенный прогресс был достигнут в 1927, когда В. Гейтлер и Ф. Лондон выполнили первый количественный квантово-химический расчёт молекулы H2. В подтверждение гипотезы Льюиса было показано, что химическая связь в H2 действительно осуществляется парой электронов и является результатом электростатического (кулоновского) взаимодействия электронов и ядер. Образование молекулы из атомов энергетически выгодно, если спины электронов направлены в противоположные стороны, когда притяжение электронов к ядру (остову) чужих атомов больше энергии отталкивания между электронами и между ядрами. Параллельная ориентация спинов приводит к отталкиванию атомов друг от друга.
В дальнейшем идеи Гейтлера - Лондона были распространены на многоатомные молекулы, что привело к созданию теории локализованных пар. Согласно этой теории, общая картина распределения электронной плотности в молекулах типа MXk складывается из независимых фрагментов М - X, связь в каждом из которых осуществлена парой электронов (по одному от центрального атома М и от заместителя X), локализованной между двумя атомами М и X. Согласно этой теории В. не просто связывается с наличием неспаренного электрона, но и характеризуется тем, в каком состоянии этот электрон находится или, в терминах теории химической связи, какую атомную орбиталь (АО) он занимает. АО разного типа имеют различную ориентацию в пространстве: s-орбиталь сферически симметрична, орбитали px, ру и pz вытянуты вдоль трёх взаимно перпендикулярных осей и т.д. Электроны атомов в молекулах в общем случае описываются "гибридными" (смешанными) орбиталями, в которые, в принципе, могут входить любые валентные АО в разных количественных соотношениях и у которых электронные облака сконцентрированы вдоль направлений связей М - Х значительно сильнее, чем у простых АО. Состояние валентных электронов, а следовательно и свойства В. атома М, в значительной мере определяют закономерности в свойствах молекул MXk для широкого круга заместителей X. Наиболее плодотворными оказались концепции направленных В. и валентных состояний атомов, позволившие объяснить и обобщить ряд закономерностей в геометрическом строении и энергиях химических связей органических и неорганических молекул.
В теории направленных валентностей предполагается, что связи М - Х в молекулах MXk тем прочнее, чем больше перекрывание электронных облаков гибридных орбиталей атомов М и X, то есть чем сильнее эти облака сконцентрированы вдоль направлений М - X. Поэтому молекулы MXk должны иметь такое геометрическое строение, при котором плотность гибридных АО вдоль направлений связей максимальна, а валентные углы Х - М - Х совпадают с углами между направлениями гибридных АО центрального атома. Например, в молекулах типа PH3 и SH2 связи осуществляются почти чистыми 3р-орбиталями центральных атомов, и поэтому PH3 и SH2 имеют пирамидальное и угловое строение с углами Н - М - Н ~ 90. В дигалогенидах Zn, Cd, Hg, двуокисях, дисульфидах и др. соединениях углерода и его аналогов связи образуются за счёт sp-гибридных АО с валентным углом 180, так что все молекулы типа CdCl2, Hg(CH3)2, HgI2, CS2, SiO2 и др. в парах имеют линейное строение. В случае Са, Sr, Вa, Ra и переходных металлов III-VI групп смешанная гибридизация sp + sd приводит к тому, что молекулы типа CaF2, SrF2, BaHal2, TiO2, HfO2, TaO2, ThO2, UO2 и др. имеют угловое строение.
С проблемой В. тесно связано приближённое понятие валентного состояния атома- гипотетического состояния, в котором находится атом в молекуле. Оно характеризуется валентной конфигурацией, то есть типом и числом заполненных и пустых валентных АО; их гибридизацией, воспроизводящей геометрическое строение ближайшего окружения рассматриваемого атома; числом электронов (в теории локализованных пар - это целое число: 2, 1 или 0), заселяющих каждую из гибридных АО, и относительной ориентацией спинов электронов. Например, в молекуле метана CH4 атом С (см. рис. 4) имеет валентную конфигурацию 2s2p3 с четырьмя тетрагональными sp3-гибридными орбиталями (te), направленными к вершинам тетраэдра, каждая из которых заселена одним электроном с неопределенно ориентированным спином, осуществляющим одну гайтлер-лондоновскую связь с соответствующим атомом Н. Как правило, валентное состояние атома в молекуле не совпадает с основным состоянием изолированного атома. Так, у углерода и его аналогов основное состояние (рис. 4, а) может быть лишь двухвалентным. У всех атомов II группы периодической системы основное состояние s2 вообще не может быть валентным, и для образования молекул типа ZnCl и ZnCl2 необходимо возбуждение s-электрона на ближайший пустой р-уровень. Энергия возбуждения валентного состояния из основного состояния для разных атомов различна и может достигать нескольких сотен ккал/моль, давая существенный вклад в общий энергетический баланс образования молекул из атомов. В случае Zn, Cd и Hg возбуждение s р происходит при присоединении первого атома галогена и требует значительных затрат энергии (90-120 ккал/моль), поэтому энергия разрыва связи М - Hal в двухатомных молекулах MHal значительно меньше, чем связи HalM - Hal в трёхатомных молекулах MHal2. У Ca, Sr, Вa, Ra затраты на возбуждение s р или s d значительно меньше (30-50 ккал/моль), и здесь энергии разрыва связей в молекулах галогенидов гораздо ближе друг другу.
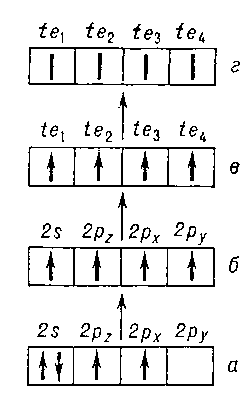
Рис. 4. Схема возбуждения валентного состояния (г) атома углерода в молекуле типа CH4 из основного состояния (а): а — основное состояние наинизшей конфигурации 2s22p2; б — нижнее состояние валентной конфигурации 2s2p3; в— гибридизация АО; г—неопределённая ориентация спинов валентных электронов (валентное состояние).
В комплексных соединениях координационное число центрального атома часто больше числа электронов в его валентной оболочке. Важную роль здесь играют донорно-акцепторная связь и дативные связи, образующиеся за счёт неподелённой электронной пары (то есть пары электронов с противоположными спинами, занимающих одну АО) одного атома и пустой орбитали другого. Соответственно должны быть расширены и представления о В.: способность к образованию связей, а следовательно и В. атома, обусловливается не только неспаренными электронами, но и неподелёнными парами и пустыми орбиталями валентной оболочки. Наибольшая суммарная В. должна быть равна числу всех АО, составляющих валентную оболочку атома, поскольку каждая валентная АО, независимо от того, сколькими электронами она заселена у атома в валентном состоянии, потенциально способна образовать одну связь (гайтлер-лондоновскую, донорно-акцепторную или дативную). В рамках этой концепции максимальная В. всех элементов второго периода от Li до F равна 4 (одна s-opбиталь + три р-орбитали), у элементов следующих периодов - 9 (за счёт ещё пяти d-opбиталей) и т.д. Решение же вопроса о том, какие из этих четырёх или девяти В. насыщаются и какие остаются неиспользованными, в соединениях каждого конкретного типа определяется не только свойствами самого атома и его положением в периодической системе, но и особенностями соединения в целом. Полный ответ на него может быть получен с помощью квантово-химических расчётов. За счёт донорно-акцепторного взаимодействия фактическое число связей атома (а следовательно и его В.) в комплексных и даже в простых соединениях в общем случае может быть больше не только числа его неспаренных электронов, но и числа связанных с ним соседних атомов.
Следует помнить, что подразделение связей в соединениях на гайтлер-лондоновские, донорно-акцепторные и дативные имеет, вообще говоря, лишь генетический смысл, поскольку после того как соединение образуется, в нём происходит перераспределение электронной плотности и выравнивание связей: например, в каждом из комплексных анионов типа [BF4]-, [BeF4]2-, [SiFe6]2-, [АlF6]3-, [ZnF6]4- и др. все связи М - F совершенно одинаковы.
Установлено также, что в солях ион NO3- имеет структуру правильного треугольника, а ионы - структуру правильного тетраэдра. Поэтому строение молекул соответствующих солей точнее описывается приведёнными на рис. 5 структурными формулами г - е, а не традиционными формулами а - в, которые не учитывают реальной структуры ионов.
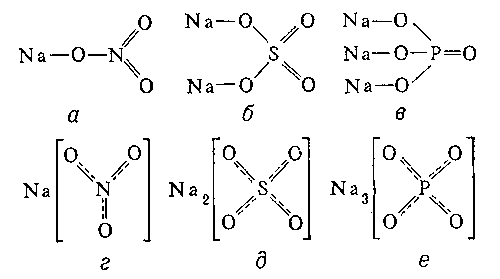
Рис. 5. Структурные формулы молекул: NaNO3 (а, г), Na2SO4 (б, д) и Na3PO4 (в, е)
Теория локализованных пар ограничена в основном несопряжёнными органическими и простыми неорганическими соединениями. Так, в случае "электронно-избыточных" молекул типа PF5, SF6, IF7, XeF6 эта теория не может объяснить осуществления высших В. у атомов Р, S, I, Xe без привлечения валентных состояний с большими целочисленными заселённостями внешних d-opбиталей (sp3d для Р, sp3d3 для I, s2p3d3 для Xe и т.д.); однако энергии возбуждения последних столь велики (200-400 ккал/моль и более), что затраты на их возбуждение вряд ли могут окупиться за счёт выигрыша в энергии при образовании связей. Аналогичные трудности возникают при рассмотрении комплексных соединений, координационных кристаллов и т.д. В "электронно-дефицитных" молекулах типа В2Н6 (рис. 1, в) число связей, образуемых атомом Н, больше числа имеющихся у него валентных АО, так что связи мостиковых Н с двумя атомами В могут быть описаны только трёхцентровыми молекулярными орбиталями, охватывающими фрагменты В - Н - В. В случае ароматических и сопряжённых молекул типа C5H5, C6H6, C7H7 и др., их комплексов с металлами (рис. 3) и других производных валентные 2рp-электроны в равной степени принадлежат всем атомам С и могут быть описаны лишь с помощью делокализованных молекулярных орбиталей, охватывающих всё кольцо или углеродный остов в целом. Иными словами, представления о локализованных В. и связях оказались слишком узкими, чтобы вместить все известные типы соединений.
Поэтому естественным следующим шагом в развитии общей теории В. стал метод молекулярных орбиталей, MO, который рассматривает молекулу как совокупность ядер и электронов, где каждый электрон движется в поле остальных электронов и всех ядер. Молекулярные орбитали, описывающие состояние электронов, в общем случае охватывают все атомы молекулы, так что каждый атом способен в принципе образовывать связи со всеми остальными атомами молекулы. Метод МО значительно более общ и последователен, что делает его в принципе пригодным для описания любых классов соединений.
Дата добавления: 2016-05-30; просмотров: 3395;
Поиск по сайту
Узнать еще
- I. Способы представления переменного синусоидального тока и напряжения.
- Античные представления о культуре
- База данных и модель представления данных
- Биологические свойства пар «цитокин — клетка–мишень» 1 глава
- Биологические свойства пар «цитокин — клетка–мишень» 2 глава
- Биологические свойства пар «цитокин — клетка–мишень» 3 глава
- Биологические свойства пар «цитокин — клетка–мишень» 4 глава
- Биологические свойства пар «цитокин — клетка–мишень» 5 глава

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
