Болезни с нетрадиционным типом наследования
Как отмечалось выше, в последние годы обнаружен ряд новых закономерностей наследования признаков (нормальных и патологических), не соответствующих менделевским. К ним относятся: геномный импринтинг, экспансия триплетных повторов, митохондриальные болезни.
Совсем недавно у человека обнаружены однородительские дисомии.В норме ребенок наследует по каждой паре хромосом: одну хромосому от отца, другую - от матери. У индивидов с этим типом
наследования число хромосом по всем парам нормальное, однако одна пара представлена хромосомами от одного и того же родителя. Наиболее частый механизм возникновения однородительской дисомии у человека - редукция трисомии: в процессе гаметогенеза за счет нерасхождения хромосом возникает дисомия в гамете по определенной хромосоме, что при оплодотворении приводит к трисомии. По неясным пока причинам третья хромосома может элиминироваться на ранних стадиях дробления, а у зародыша останутся две хромосомы одного родителя. Следует отметить, что в целом частота возникновения однородительской дисомии материнского происхождения в 3 раза чаще отцовской. Если бы две копии генов, получаемых потомками от матери или от отца, функционировали в клетках одинаково, то никаких серьезных последствий в организме эти нарушения не вызывали бы. Однако оказалось, что некоторые хромосомы несут отдельные гены, экспрессия которых определяется полом передавшего их родителя.Это явление получило название феномена геномного импринтинга (от imprint - отпечаток).
Под геномным импринтингом понимают эпигенетический процесс, который дифференциально маркирует материнские и отцовские гомологичные хромосомы. В участках генома, подверженных импринтингу, экспрессируется (проявляется) только один из двух аллелей - отцовский или материнский, т.е. наблюдается моноаллельная экспрессия генов. Второй аллель, вследствие наличия на нем некоего отпечатка, импринтирован (выключен или подавлен) и не экспрессируется.
Такой способ регуляции генов свидетельствует о неэквивалентном вкладе родителей в геном потомков и приводит к разному фенотипическому проявлению мутаций у потомства, унаследованных от матери или отца.
У человека известно уже около 40 таких генов, и предполагается, что их число может достигать 200-500. Основную роль в процессе геномного импринтинга играет метилирование цитозиновых оснований ДНК с образованием 5-метилцитозина, способствующее выключению экспрессии генов с модифицированными нуклеотидами.
Так, известно, что в проксимальном участке хромосомы 15 имеются близко сцепленные, но противоположно импринтированные локусы, отвечающие за возникновение двух фенотипически разных синдромов - Прадера-Вилли и Энгельмана. Для синдрома
Прадера-Вилли,фенотипически проявляющегося умственной отсталостью, мышечной гипотонией, выраженным ожирением, гипогонадизмом, низким ростом и акромикрией, установлен кандидатный ген, ответственный за синтез полипептида N малого ядерного рибонуклеопротеина (SNRPN). Этот ген активно экспрессируется исключительно на отцовской хромосоме 15. Кандидатным геномом синдрома Энгельмана(синоним - синдром «счастливой куклы»), характеризующегося неадекватной счастливой улыбкой и глубокой умственной отсталостью с резкими кукольными судорожными движениями, является убиквитин - белковый лигазный ген (ИВЕЗА) E6-AP, который экспрессируется главным образом на материнской хромосоме 15.
Синдром Прадера-Виллиразвивается при делеции участка 15 хромосомы отца (нет участка, который экспрессируется на отцовской хромосоме) или однородительской дисомии 15 хромосомы материнского происхождения (в этом случае гены импринтированы - выключены или подавлены).
Синдром Энгельмана («счастливой куклы»)развивается при делеции участка 15 хромосомы матери или при однородительской дисомии 15 хромосомы отцовского происхождения.
В случае однородительской дисомии отсутствует потеря хромосомного материала, но нарушается нормальное метилирование ДНК, а следовательно, и транскрипция генов критического района, поэтому такое функциональное состояние равнозначно делеции.
В начале 1990-х гг. у человека был обнаружен новый тип мутаций, который до сих пор не зарегистрирован ни у одного вида млекопитающих. Он получил название динамических мутаций или мутаций экспансии(см. выше).
Общие характеристики этого класса болезней следующие:
1. Болезни с экспансией тринуклеотидных повторов представляют собой нейродегенеративные заболевания с поздним проявлением.
2. Отмечается прямая корреляция между числом тринуклеотидных повторов и тяжестью клинической картины.
3. Для болезней экспансии характерна генетическая антиципация- возрастание тяжести заболевания в последующих поколениях, что связано с тенденцией к возрастанию числа повторов у потомков.
Первое заболевание, при исследовании которого в 1991 г. был открыт феномен экспансии - синдром фрагильной (ломкой)
Х-хромосомы,или синдром Мартин-Белл. Проявления синдрома Мартин-Белл: умственная отсталость, аутизм, макроорхидизм (у взрослых), удлиненное лицо, прогнатия - выступающий подбородок, оттопыренные уши, пронзительная смешная речь, аномалии соединительной ткани, нарушение поведения.
Сейчас открыта целая группа болезней экспансии тринуклеотидных повторов.
Утверждение, что весь генетический материал человека находится в составе хромосом, не совсем верно, поскольку есть одно исключение - митохондриальный геном.Митохондриальная ДНК (мтДНК) представляет собой небольшую кольцевидную молекулу длиной 16 569 пар оснований. В отличие от ДНК ядерного генома она не связана с белками, имеет очень высокую «плотность генов» ввиду отсутствия интронов, содержит 13 генов, кодирующих белки (3 субъединицы цитохром-С-оксидазы, 6 компонентов АТФазы и др.), 22 гена транспортных РНК (тРНК) и 2 гена рибосомальных РНК. На рисунке 5-11 представлена схема структуры мтДНК и приведены примеры митохондриальных болезней, которые являются следствием мутации мт-генов. Эти болезни передаются только по материнской линии.
Наследование признаков, передаваемых через ДНК митохондрий, и связь мутаций мтДНК с болезнями человека были впервые показаны в 1988 г. С тех пор обнаружено большое число мутаций мтДНК, лежащих в основе целого ряда нейродегенеративных заболеваний, некоторых МФЗ, митохондриальных миопатий.
Примеры таких заболеваний:
• синдром MELAS, развивающийся в связи с мутацией в гене лейциновой тРНК митохондрий и проявляющийся митохондриальной энцефаломиопатией, лактатацидозом и инсультоподобными эпизодами. Вначале развивается сахарный диабет 2 типа с инсулинорезистентностью, который переходит в сахарный диабет 1 типа с инсулинонедостаточностью и наличием аутоантител к антигенам островков поджелудочной железы;
• наследственная нейроофтальмия Лебера (характеризуется билатеральной потерей зрения);
• синдром MERRF (характеризуется прогрессирующей дегенерацией нервной и мышечной ткани, что проявляется судорогами, атаксией, миопатией, потерей слуха);
• летальная инфантильная дыхательная недостаточность и др.
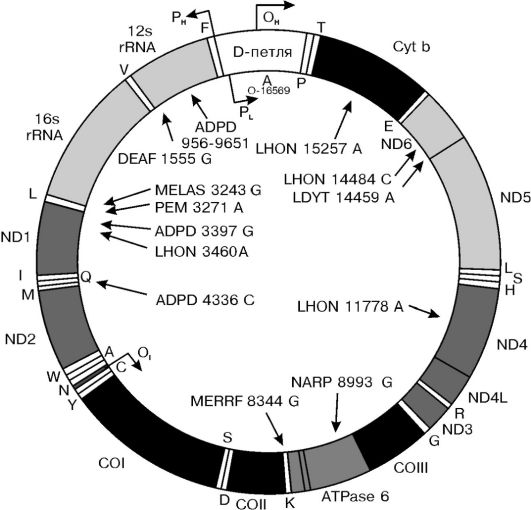 Рис. 5-11. Структура митохондриального генома и примеры митохондриальных болезней: APDP - болезнь Альцгеймера/болезнь Паркинсона; DEAF - нейросенсорная потеря слуха; LHON - наследственная нейроофтальмия Лебера; LDYT - LHON и дистония; MELAS - митохондриальная миотония, энцефалопатия, молочнокислый ацидоз и приступы судорог; MERRF - миоклональная эпилепсия в сочетании с необычно красными мышечными волокнами; NARP - нейропатия, атаксия и пигментный ретинит; PEM - летальная прогрессирующая энцефаломиопатия
Рис. 5-11. Структура митохондриального генома и примеры митохондриальных болезней: APDP - болезнь Альцгеймера/болезнь Паркинсона; DEAF - нейросенсорная потеря слуха; LHON - наследственная нейроофтальмия Лебера; LDYT - LHON и дистония; MELAS - митохондриальная миотония, энцефалопатия, молочнокислый ацидоз и приступы судорог; MERRF - миоклональная эпилепсия в сочетании с необычно красными мышечными волокнами; NARP - нейропатия, атаксия и пигментный ретинит; PEM - летальная прогрессирующая энцефаломиопатия
Дата добавления: 2016-07-11; просмотров: 3950;
Поиск по сайту
Узнать еще
- Аллергические болезни
- Артериосклероз. Атеросклероз. Артериальная гипертензия: гипертоническая болезнь и вторичные артериальные гипертензии. Ишемические болезни сердца (ИБС)
- Аутосомно-доминантный тип наследования
- Аутосомно-рецессивные болезни
- Аутосомно-рецессивный тип наследования
- Бактерии и болезни, вызываемые бактериями
- Болезни верхних дыхательных путей
- БОЛЕЗНИ ВЗРОСЛЫХ НАСАЖДЕНИЙ

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
