Аутосомно-рецессивные болезни
| Болезяи | Проявление |
| Серповидно-клеточная анемия | Развитие хронической гипоксии и анемии с расстройствами кровообращения и тромбозами |
| Цистический фиброз | Нарушения функций поджелудочной и других желез, образование толстого слоя слизи, ведущей к пневмонии и эмфиземе. Смерть наступает обычно в детском возрасте. Представляет собой наиболее частую генетическую аномалию детей (1 на 3700 рождений) |
| Галактоземия | Пониженные уровни галактозо-1-фосфатуридилтранс-феразы ведут к увеличению печени, катаракте и нарушениям психики |
| Гидроцефалия | Избыточное накопление жидкости в черепной коробке, вызывающее физические и психические нарушения |
| Глухота врожденная | Около половины случаев детской глухоты вызывается этим аллелем |
| Болезнь Нимена-Пика | Накопление липидов в нервных клетках, вызывающее нарушение психики, увеличение печени, замедленный рост. Смерть наступает обычно в первые три года жизни |
| Пернициозная анемия | Нарушения в метаболизме витамина Вдг, что ведет к симптомам, связанным с уменьшением в крови количества эритроцитов |
| Болезнь Тея-Са-са | Прогрессивно развивающиеся паралитические явления, нарушения психики, слепота. Смерть наступает в первые три года жизни. В 27—53% случаев встречается в браках двоюродных сестер и братьев |
| Фенилкетонурия | Нарушения в тонусе мышц, уменьшение пигментации кожи, волос, радужной оболочки глаз, микроцефалия, умственная отсталость |
Аутосомно-рецессивные болезни (табл. 27), в общем, встречаются чаще, чем аутосомно-доминантные, и проявляются лишь у гомозиготных носителей мутантных аллелей, рождающихся в семьях, где оба родителя являются гетерозиготными или один является гомозиготой, второй — гетерозиготой. Конечно, больные рождаются и в семьях, где оба родителя являются гомозиготными носителями мутантных аллелей (больными).
Для распространения болезней, наследуемых по этому типу, характерна неравномерность. Например, такие болезни, как болезни Тея-Сакса, Нимена-Пика и мышечная деформирующая дистония очень часты среди восточно-европейских евреев (ашкенази), достигая частот, превышающих частоты этих болезней среди населения других национальных групп. Например, болезнь Тея-Сакса, причиной которой является мутация гена, контролирующего лизосом-ную гексозоаминидазу А, среди евреев-ашкенази в Австрии составляет 11%, в Чехии и Словакии — 9%, в Венгрии — 7%, в Румынии — 4%, в Польше, бывшем СССР — 3%. Однако среди людей этой группы очень редка фенилкетонурия. Акаталазия, наследственный дисхроматоз и некоторые другие болезни с высокой частотой встречаются среди японцев. Кровные браки способствуют появлению гомозиготных рецессивных носителей очень редких му-тантных аллелей, и в этом состоит большая генетическая опасность кровных браков.
Еще в начале века было показано, что эритроциты многих индивидуумов агглютинируются сывороткой кроликов, иммунизированных кровью обезьян-резус. Эритроцитарный антиген, который ответственен за эту реакцию, получил название резус-фактора, а ген, который детерминирует это свойство, был назван Rr (или Rh rh). Следовательно, индивидуумы, эритроциты которых содержат Rh-фактор, являются резус-положительными (Rh+). У резус-положительных индивидуумов (Rh+) отец также является Rh+ (генотип RR), но мать Rh- (генотип rr). Когда резус-положительный плод находится в матке резус-негативной женщины (рис. 158), то он вызывает образование в крови матери Rh-антител, которые реагируют с эритроцитами плода. Эта реакция сопровождается развитием анемии плода и его абортом или смертью после рождения.
Такая аутосомно-рецессив 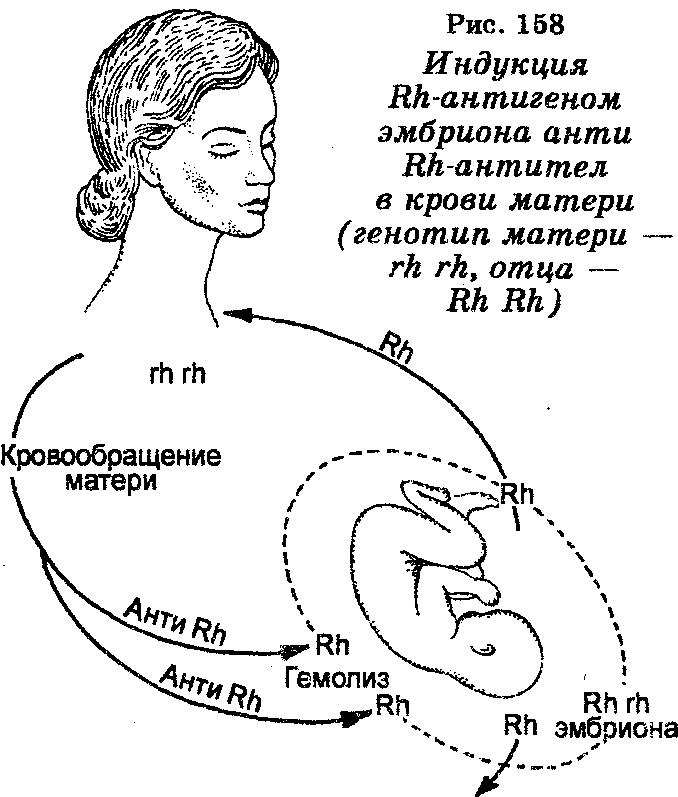 ная аномалия, как альбинизм, который обусловлен рецессивным геном, вследствие чего альбинизм является рецессивным по отношению к нормальной пигментации, также встречается в разных странах с разной частотой. В Европе и США один альбинос приходится на 20 000 жителей, тогда как в Южной Америке и Африке — значительно чаще.
ная аномалия, как альбинизм, который обусловлен рецессивным геном, вследствие чего альбинизм является рецессивным по отношению к нормальной пигментации, также встречается в разных странах с разной частотой. В Европе и США один альбинос приходится на 20 000 жителей, тогда как в Южной Америке и Африке — значительно чаще.
Анализ родословных многих семей, в которых обнаружены альбиносы, свидетельствует, что дети-альбиносы обычно рождаются от родителей-неальбиносов наряду с детьми-неальбиносами, причем отношение между детьми-неальбиносами и детьми-альбиносами является менделевским (3 : 1). Решающие доказательства того, что альбинизм — это наследственный признак, получены при изучении идентичных близнецов. Как правило, если один из них альбинос, другой также является альбиносом, тогда как из двух неидентичных близнецов лишь один может быть альбиносом. Неальбиносы являются либо гомозиготными доминантными организмами (АА), или гетерозиготными организмами (Аа). Альбиносы являются гомозиготными рецессивными организмами (аа). Брак между альбиносом и неальбиносом не дает детей-альбиносов, т.к. рецессивный аллель а редок и большинство людей не являются носителями такого аллеля. В браке между альбиносом и неальбиносом дети-альбиносы могут быть только тогда, когда неальбинос является гетерозиготным организмом, причем среди детей соотношение между альбиносами и неальбиносами будет составлять 1 : 1. В браках между альбиносами рождающиеся дети всегда альбиносы.
Среди наследственных болезней, передающихся в качестве ауто-сомно-рецессивных признаков и приуроченных к отдельным географическим районам, интерес представляет серповидно-клеточная анемия. Известно, что нормальные эритроциты обладают гемоглобином А, каждая молекула которого состоит из двух поли-пептидных цепей к и двух цепей Р. Каждый й-полипептид представлен специфическими последовательностями, состоящими из 141 аминокислоты, а каждый а-полипептид — специфическими последовательностями, состоящими из 149 аминокислот. Напротив, у больных серповидно-клеточной анемией были найдены эритроциты серповидной формы, которые обладают гемоглобином S, являющимся результатом мутации (замены пары А-Т на пару Т-А) гена НbА, контролирующего синтез гемоглобина А, к аллелю НbbS, контролирующему синтез гемоглобина S. Данная мутация сопровождается тем, что в эритроцитах больных одна половина гемоглобина оказывается гемоглобином А, другая половина — гемоглобином S. Химические различия между гемоглобинами А и S заключаются в том, что b-полипептидные цепи в их глобинах (белковых частях) различаются одной аминокислотной заменой: в b-цепи гемоглобина А шестой аминокислотой является глутаминовая кислота, а в b-цепи гемоглобина S — валин. Генотип индивидов, гомозиготных по S-аллелю, будет HbbS/ HbbS. Удлиненные серповидные эритроциты не способны обеспечивать транспорт кислорода к тканям, поэтому во многих случаях болезнь заканчивается смертельно еще в детском возрасте. В результате мутаций гена HbA в виде замены в нем пар азотистых оснований происходит образование и других вариантов гемоглобина (помимо гемоглобина S), которых известно более 100 и которые обнаружены в равных популяциях людей. Эти варианты различаются между собой элек-трофоретически, а образование некоторых из них также сопровождается неблагоприятными фенотипическими эффектами разной тяжести.
Свыше 200 генов локализовано на половых хромосомах. Перечень некоторых наследственных болезней, которые контролируются генами, локализованными на Х- или Y-хромосомах, приведен в табл. 28. На хромосоме Х идентифицировано свыше 70 генов, контролирующих гемофилию, мышечную дистрофию, цветовую слепоту (дальтонизм), ювенильную глаукому, оптическую атрофию (дегенерацию зрительного нерва) и др. Большинство из этих болезней наследуются по рецессивному типу. Доминантный тип наследования болезней, которые детерминируются генами, сцепленными о Х-хромосомой, очень редок.
Таблица 28
Дата добавления: 2016-05-30; просмотров: 3107;
Поиск по сайту
Узнать еще
- Аллергические болезни
- Артериосклероз. Атеросклероз. Артериальная гипертензия: гипертоническая болезнь и вторичные артериальные гипертензии. Ишемические болезни сердца (ИБС)
- Бактерии и болезни, вызываемые бактериями
- Болезни верхних дыхательных путей
- БОЛЕЗНИ ВЗРОСЛЫХ НАСАЖДЕНИЙ
- Болезни гемоглобинов
- Болезни глаза и придаточного аппарата

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
