Азатиоприн Аллопуринол
149. Контрастные средства: иодсодержащие рентгеноконтрастные средства (кислота амидотризоевая); контрастные средства для МРТ (кислота гадопентатовая, гадодиамид); контрастные средства для ультразвукового исследования (галактоза). Химическое строение. Связь структуры и действия.Рентгеноконтрастные средства (РКС) –группа контрастных средств, применяемых в рентгенодиагностике.
 Амидотризоевая кислота Водорастворимое нефротропное высокоосмолярное контрастная среда.
Амидотризоевая кислота Водорастворимое нефротропное высокоосмолярное контрастная среда.
Свойства идеального РКС: растворимость в воде, химическая и термическая стабильность; биологическая инертность; низкая вязкость; осмоляльность; селективная экскреция; безопасность; низкая стоимость.
В качестве КС для МРТ применяют соединения Gd(III). Атомы гадолиния
 |

|
Ультразвук – звуковые колебания, имеющие частоту более 20 кГц. Контрастные вещества – Микроцастицы галактозы.
 При суспендировании микрогранул галактозы (частиц размером меньше 1 мкм) на их поверхности адсорбируется воздух, который при соприкосновении с тканями высвобождается в виде пузырьков. Эти пузырьки усиливают амплитуду отраженного сигнала, повышая контрастность изображения при эхографиии УЗИ
При суспендировании микрогранул галактозы (частиц размером меньше 1 мкм) на их поверхности адсорбируется воздух, который при соприкосновении с тканями высвобождается в виде пузырьков. Эти пузырьки усиливают амплитуду отраженного сигнала, повышая контрастность изображения при эхографиии УЗИ
150. Радиофармацевтические ср-ва.: соед. Технеция(99mTc), натрия йодогиппурат(131J), натрия фосфат(32Р), стронция(89Sr )хлорид…
Способн. излучения радиоакт. изотопов(РИ) проникать сквозь в-во,
ионизировать его, воздейств. на ткани живого орг-ма, а также разнообразие самих РИи их физич. характеристик создает широк. возможн. для использ-я их в науке и практике.
В мед. РИиспольз. по 2 направлениям:
- РИ с малой активностью излучения– в кач-ве «меченых» атомов (для диагн. целей, для изуч. фармакол. д-я лек. ср-ва, в установл. скорости его проникновения, локализ., выведения из орг-ма),
- РИ с > активностью излуч.– в кач-ве носителей излучения для лечения
гл. образом злокач. новообразований.
Для медицины >
интерес представляют РИфосфора -32Р, натрия -24Nа,
йода - 131Í,хрома 51Сr, технеция -99mTc, кобальта-29Со…
32Р прим. для леч. лейкемии. Он также использ. для изуч. п-сов фосфорного обмена в различн. клетках, органах и тканях.
24Nа использ. в хирургии для определ. скорости кровотока, зоны нарушен. кровообращения.
Радиоакт. 131Jприм. при расстройствах ЩЖ при гипертиреоидии. В завис.
от состояния щитовидн. ж-зы в ней фиксир-ся этим 131Jшироко прим. в диагностич. целях.
> или <
колич-во йода. В связи с
Радиоакт. кобальтисп. в кач-ве источн. жесткого излучения ( γ -пушка),
радиоакт. технецийдля изуч.я фармакологии и фармакодинамики в тканях организма.
Натрия хромат, меченый 51Сr, прим. для диагн. целей при различн. заболев. крови и для диагн. желуд.-кишеч. кровотечений.
При работе с радиоактивными препаратами необходима соответствующая защита от излучения этих препаратов. Защита от внешнего α- и β-излучения РИосущ. просто из-за их малой проникающ. способн. Пробег α-частиц в воздухе не
> неск. см. α-частицы поглощ-ся резин. перчатками, одеждой, стенками стекл.
ампулы. Пробег β-частиц в воздухе в завис. от их энергии = от неск. см до неск. метров. Для защиты от β-излуч. прим. материалы с малой атомн. массой = спец. экраны из оргстекла, контейнеры из алюминия, пластмассы. Источн. жесткого β-излуч. экранируют свинцом. γ- излуч. в отличие от α- и β- не характеризуется опред. пробегом в в-ве – оно поглощ-ся по мере прохожд. ч/з в-во по экспоненциальному закону. Наиб. эффективно поглощ. γ- излуч. в-ва с большими атомн. массами, напр. свинец, золото и др.
 |
198. Общая характеристика способов получения антибиотиков (АБ.).Различают три возможных способа получения АБ: биосинтез;
химическая или биотехнологическая модификация природных АБ. и химический синтез. АБ. обладает сложной химической структурой, поэтому их полный химический синтез очень трудоёмок и экономически невыгоден. Исключение составляют хлорамфеникол и др. в-ва, имеющие простое химическое строение.
Основным путём получения АБ. является биотехнологический способ. АБ. продуцируются плесневыми грибами, актиномицетами, эубактериями и др. микроорганизмами. (Пенициллин - Penicillium chrysogenum, P. Notatum; Цефалоспорин - Cephalosporum acremonium; Стрептомицин - Streptomyces globisporus streptomycini и др.) Один и тот же вид микроорганизмов может синтезировать несколько АБ. Напр., Streptomyces griseus синтезирует более 50 АБ.
Варианты биотехнологического получения природных и полусинтетических АБ.
1. Прямая ферментация микроорганизма-продуцента с в-вом, явл. метаболическим предшественником получаемого АБ и стимулирующим процесс его биосинтеза. Напр, биосинтез бензилпенициллина ведут в присутствии фенилуксусной кислоты, макролидов – в присутствии пропионовой кислоты и пропилового спирта.
2. Использование для биосинтеза АБ. микроорганизмов-мутантов, у которых блокированы определённые ферменты, участвующие в синтезе АБ. Если в среду, содержащую такой микроорганизм ввести аналог предшественника АБ, то получиться модифицированный АБ. (исп. для получения полусинтетич. пенициллинов и цефалоспоринов.
Большинство АБ получают при глубинной аэробной ферментации периодического действия в асептических условиях.
Процесс биосинтеза АБ состоит из 2 этапов:
1. Накопление достаточного количества биомассы, которая выращивается на среде для роста микроорганизмов. Должен протекать быстро, а пит. среда д. б. дешёвой.
2. Активный синтез АБ. Ферментацию ведут на продуктивной среде. Т.к. АБ явл.
вторичными метаболитами, их биосинтез происходит в стационарной фазе (идиофазе). Любые механизмы, тормозящие пролиферацию и активный рост, активируют процесс образования АБ.
Завершающими этапами получения АБ явл. стадии выделения и очистки. Данные процессы определяются природой АБ, характером производства и целями дальнейшего использования антибиотиков. Для выделения и очистки АБ применяют методы:
*экстракция органическими растворителями;
*сорбция;
*осаждение и перекристаллизация из различных сред;
*ионообменная хроматография и др.
Выделенные и очищенные АБ подвергают лиофильной и распылительной сушке.
202.Общ. х-ка м-дов оценки кач-ва антибиотиков (АБ).М-ды контроля кач-ва:
1.Спектрометрические (спектральные х-ки: поглощение УФ- и ИК- излучения; спектрофотометрия; ЯМР-спектроскопия)
2.Хроматографические (ТСХ, ВЭЖХ, ГЖХ, бумажная)
3.Химические (разл. химич. реакции)
4.Биологические-напр. микробиологич. м-д колич. определения АБ.
5.Другие.
№4:Определяют путем сравнения степени угнетения роста чувствительных МО под д-ем испытуемого АБ и стандартного образца в известных концентрациях. Сущ. 2 способа:
А) М-д диффузии:-проводят на твердых средах, их засевают определенным колич-вом указанных в НД тест-МО (микроорганизмов). На поверхн. среды наносят р-р исследуемого АБ и стандартного образца(СО). Инкубируют определенное время. Измеряют диаметр зон угнетения роста тест-МО, вызванного исследуемым АБ и стандартн. образцом.
Б) Турбидиметрический м-д:из растворителя и буферного р-ра (указ. в ГФ РБ) готовят р-ры стандартного образца и испытуемого АБ, имеющие известные концентрации и предположительно равные активности. Помещают равные объемы каждого р-ра в идентич. пробирки, + в каждую равные объемы инокулированной среды( напр. 1мл р-ра + 9мл среды). Инкубируем. Затем останавливаем рост МО добавлением 0,5мл р-ра формальдегида в каждую пробирку, или тепловой обработкой. Измеряют степень мутности.
Для характеристики колич. содержания действ. в-ва в образце АБ, кроме обычных параметров используют ЕД. ЕД-минимальная масса АБ, которая подавляет развитие тест-микроорганизма в определенном объеме питательной среды. Обычно 1 ЕД соответствует 1 мкг чистого АБ(стрептомицин, тетрациклин), но 1 ЕД натриевой соли бензилпенициллина соотв. 0,5958 мкг данного в-ва.
 |  |

 203.Способы пол-я и МКК пенициллинов
203.Способы пол-я и МКК пенициллинов
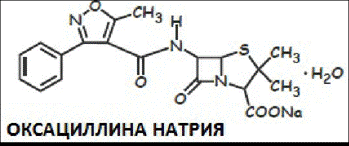
 |
МКК:1.идентиф.(ИК,УФ, ТСХ) 2.колич.опред
Кол. Опр-е проводится м-ом ВЭЖХ, спектрофотометрически по реакции с имидазола ртути (2) хлоридом
 |
204. Способы получения и методы контроля качества цефалоспоринов.
Способы получения.Исх. в-вом для синтеза является 7-АЦК, которую получают из цефалоспорина С, синтезируемого БТ способом. В отличие от 6- АПК 7-АЦК сложно получить ферментативным гидролизом, основным методом получения 7-АЦК является химическое дезацилирование. При получении ЦС проводят ацилирование 7-АЦК по аминогруппе и модификацию ацетилоксиметильной группы.Внешний вид. Большинство цефалоспоринов представляют собой натриевые соли соответствующих кислот. Данные вещества являются белыми или почти белыми порошками (некоторые могут быть желтоватыми), которые легко р-римы в воде. Некоторые цефалоспорины (особенно цефазолина натриевая соль) гигроскопичны. Цефалексин, цефаклор и натриевые соли цефтриаксона и цефтазидима являются кристаллогидратами. Кислотные формы цефалоспоринов (цефалексин, цефтазидим) и эфиры (цефуроксима аксетил) мало р-римы в воде. В этиловом спирте как солевые, так и кислотные формы антибиотиков данной группы растворяются хуже, чем в воде. Все формы цефалоспоринов практически нерастворимы в хлороформе и эфире.Спектральные свойства. Поглощение УФ-излучения. Цефалоспорины поглощают электромагнитное излучение ближнего УФ-диапазона, что обусловлено присутствием в молекулах данных веществ хромофора, состоящего из двойной связи C=C, сопряжённой с амидной группой.
Поглощение ИК-излучения. важнейшие полосы поглощения цефалоспоринов в
ИК-спектре находятся в диапазоне 1800–1500 см-1. Оптическая активность.В отличие от пенициллинов цефалоспорины могут быть как правовращающими, так и левовращающими веществами. Так натриевые соли цефотаксима, цефуроксима, а также цефалексин вращают плоскость поляризации плоскополяризованного света вправо, в то время как натриевые соли цефазолина и цефтриаксона – влево. Абсолютные величины удельного вращения у цефалоспоринов меньше, чем у пенициллинов.Идентификация.ИК- спектроскопия и, в случае натриевых солей, проба на присутствие катиона натрия. Идентификацию некоторых цефалоспоринов проводят методом ТСХ. В качестве неподв. фазы исп. силанизированный силикагель, содержащий флуоресцентный индикатор. Подв. фаза – смесь орг-ского р-рителя (ацетонитрил, ацетон, тетрагидрофуран, метилацетат) и водного р-ра ацетата аммония. Обнаружение пятен веществ проводят с помощью УФ-лампы. При идентификации некоторых цефалоспоринов исп. также реакция с реактивом Марки и индивидуальные реакции. Испытания на чистоту.При испытаниях у цефалоспоринов определяются прозрачность и цветность, рН, удельное вращение, оптическая плотность, сопутствующие примеси, содержание остаточных растворителей (N,N-диметиланилин, 2-этилгексановая кислота), вода, стерильность, бактериальные эндотоксины, сульфатная зола и тяжёлые металлы; остаточные кол-ва орг. р-рителей и микробиол. чистоту. Так, р-ры большинства цефалоспоринов имеют слабокислую или нейтр. среду (наименьшее допустимое значение рН у цефтазидима натриевой соли, в структуре кот-рого имеется дополнительная карбоксильная группа; наибольшее
– у цефуроксима натриевой соли), содержание воды минимально у
цефуроксима аксетила, максимально – цефтазидима натриевой соли. Количественное определение. ВЭЖХ. В качестве неподв. фазы - октадецилсиликагель (большинство цефалоспоринов), а также гексилсиликагель (цефуроксим, цефиазидим) и триметилсилилсиликагель
(цефуроксим аксетил). Подв. фазы представляют собой смеси ацетонитрила или (и) метанола и водных буферных растворов (фосфатный, ацетатный, цитратный). Детекция - спектрофотометрическая. Для кол-ного опр-я цефалоспоринов исп. также УФ-спектрофометрия, кислотно-основное титрование в неводных средах.
205.Способы получения и методы контроля качества карбопенемов (К), монобактамов (М) и ингибиторов бета-лактомаз (ИβЛ).
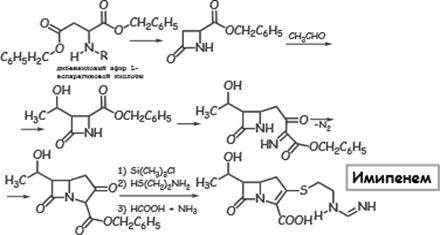
спектроскопия (спектр водного р-ра с конц. 30 мкг/мл).
К.О.
Природные Ксинтез- ся МО в незначительных кол-вах и очень неустойчивы, поэтому имипенем и меропенем получ. путём полного хим. синтеза.
Идентификация.
Проводится методом ИК-спектроскопии. Для меропенема, согл. USP, исп-ся также УФ-
Проводится методом ВЭЖХ. В кач. неподвижной фазы исп. октадецилсиликагель. Подвижная фаза для имипенема (Ph.Eur 4) – смесь (0,7:99,3) ацетонитрила и раствора K2HPO4, доведенного до рН 7,3 Н3РО4 (в USP24 в кач. подвижной фазы исп. фосфатный буфер рН 6,8 без добавления ацетонитрила); для меропенема в кач. подвижной фазы исп-ется смесь (5:1) водного р-ра триэтиламина (1,0 мл в 1 л), доведенного до рН 5,0 Н3РО4, и метанола. Детекция спектрофотометрическая (имипенем– 254 нм, меропенем– 300 нм).
М. –а/б, явл-еся производными 3-аминомонобактамовой к-ты.
 Продуценты М.- бактерии родов Acetobacterium, Gluconobacterium , Chromobacterium, отдельные виды рода Pseudomonas. Считалось, что антимикробной активностью могут обл- ть только бициклические b-лактамы.
Продуценты М.- бактерии родов Acetobacterium, Gluconobacterium , Chromobacterium, отдельные виды рода Pseudomonas. Считалось, что антимикробной активностью могут обл- ть только бициклические b-лактамы.
Практич. применение имеет лишь один представитель М– Азтреонам (получают путём полного хим. синтеза!). Для идентификацииазтреонама исп-ется ИК- спектроскопия, азтреонама для инъекций – ВЭЖХ (параллельно с колич-ым
 определением). К. О.ВЭЖХ. Неподвижная фаза – октадецилсиликагель; подвижная – смесь (4:1) фосфатного буфера (дигидрофосфат калия– Н3РО4, рН 3,0) и метанола; λ детектора – 270 нм. В случае азтреонама для инъекций в кач. неподвижной фазы исп-ся силикагель с привитыми дигидроксипропановыми
определением). К. О.ВЭЖХ. Неподвижная фаза – октадецилсиликагель; подвижная – смесь (4:1) фосфатного буфера (дигидрофосфат калия– Н3РО4, рН 3,0) и метанола; λ детектора – 270 нм. В случае азтреонама для инъекций в кач. неподвижной фазы исп-ся силикагель с привитыми дигидроксипропановыми
группами (доп. исп-ся предколонка, содерж. силикагель). Подвижная фаза
–смесь (750:250) фосфатного буфера(дигидрофосфат аммония– Н3РО4, рН 2,0) и ацетонитрила. Детектирование проводится при λ 206 нм.
Идентификация ИβЛ.Для идентификации калия клавуланата в Ph.Eur 4 исп-ется ИК-спектроскопия. В USP для идент-ции калия клавуланата и сульбактама натриевой соли применяется ВЭЖХ. Кроме того, для обоих в-в проводятся р-ции
на соотв-щие катионы. К.О.Для количественного определения обоих в-в исп-ется ВЭЖХ. Неподвижная фаза- октадецилсиликагель; подвижная для калия клавуланата – смесь (5:95) метанола и фосфатного буфера (рН4,0 - Ph.Eur и рН4,4
– USP); для сульбактама натриевой соли – смесь (1650:350) 0,005 М тетрабутиламмония гидроксида и метанола. Детекция– 230 нм.
206. Методы контроля качества аминогликозидов.(АГ)

 Амикацина(А), гентамицина(Г)и стрептомицина(С)сульфаты; тобрамицин(Т).
Амикацина(А), гентамицина(Г)и стрептомицина(С)сульфаты; тобрамицин(Т).
Все-белые в-ва, сод. много ОН-групп= р-мы в воде, нет- в этил. спирте, ацетоне, хлороформе.
 Оптич. активны- С-вращ. плоскость поляризации влево, ост. –вправо.
Оптич. активны- С-вращ. плоскость поляризации влево, ост. –вправо.
Хим. активн. обусл. гликозидн. связями(кислот. гидролиз), углеводн.
Остатками (дегидратация →
образ. замещенных фурфуро
-
лов), аминогруппами, остатками гуанидина (основные св-ва, взаимод. с общеалкалоидн. реактивами, нингидрином).
Нагрев. в щел. среде= гидролиз гуаниднновых групп= NH3↑.
При взаимод. с α-нафтолом в щел. среде→ красн. окраш.(р-я Сакагучи): Альдегидн. группа: взаимод. с окис-лителями(р-в Неслера, Толенса, Фели нга) черн.↓ в послед. р-ции; фенолами, 2,4-динитрофенил гидразном(желт.↓) Сульфат-ион:+р-мые соли бария= BaSO4↓белый, не р-м в р-рах кислот и щел)= р-я исп. для идентиф. сульфатов АГи колич. опр.(сульфатометрич. титрован.) Идентиф:ТСХ и хим. р-ции(выше), еще для Т.-спектрофотометрия ЯМР. ИК- спектроскопия- только для А., ВЭЖХ-для Г.
При опред. чистотыопред. прозрачн. и цветность ихр-ров, РН, удельн. вращение, ∆массы при высушив., сульфатн. зола, стерильн., пирогенность.
Колич. опред.-микробиологически т.к. не поглощают в ближнем УФдиапазоне= не опред-ся м-дами УФ-спектро фотометрии или ВЭЖХ со спектрофотометрич. детектированием. Для ВЭЖХ возм. превращ. АГв окраш. или флуоресцирующие соединения перед введением в хроматограф или после выхода из хроматогр. колонки. Возм ГЖХ-определение, но перед введ. в испаритель хроматографа АГпревр. в летучие триметилсилильные производные. Т.опред. ВЭЖХ с амперометрич. детектированием.
Хранят АГпо списку Б в сух., защ. от света месте, в плотно укупор. таре, при комн. температуре.
209. Методы контроля качества тетрациклинов (тетрациклин, доксициклин).
Тетрациклины (Т.) –не имеющие запаха желтые кристалл. вещ-ва.
Основания Т мало растворимы в воде, ионные формы (гидрохлориды) растворимы или легко растворимы.
Растворимость в этаноле у оснований Т. выше, чем в воде, у гидрохлоридов – наоборот.
Идентификация.1. Спектрофотометрия (УФ, реже ИК). Чаще всего
идентификацию проводят в ра-рах кислот, т.к. в растворах щелочей быстро изомеризуются. В диапазоне 200-400 нм Т.две полосы поглощения (тетрациклин в 0,05 М H2SO4 – 270 и 356 нм). 2. ТСХ. На силикогеле, предварителньо обработанном ЭДТА. Подвижная фаза – ацетонитрил:метанол:щавелевая к-та (20:20:60). Проявление: в УФ- свете при длине волны 254 нм. 3. Реакция с конц. серной к-той. Появляется фиолетово-красное окрашивание, переходящее в желтое при добавлении в воду (тетрациклин). Доксициклин дает с H2SO4 желтое окрашивание. 4. Обнаружение хлорид-ионов.
Чистота. 1. рН растворов. 2. Удельное оптическое вращение (Т.хиральны. При измерении оптической активности используют водные или метанольные ра-ры HCl. Все Т.левавращающие) 3. Сопутствующие примеси – ВЭЖХ. Специфические примеси Т.– эпи-, ангидро-, эпиангидротетрациклины. 4. Тяжелые металлы. 5. Сульфатная зола. 6. Потеря в массе при высушивании.7. Остаточные ко-ва орг. растворителей.
Количественное определение. ВЭЖХ.
210 Способы получения и методы контроля качества хлорамфеникола и его эфиров.
 |  |
В отличие от большинства других антибиотиков левомицетин получают в промышленных масштабах путём полного химического синтеза. Получение п- нитроацетофенона
1. Получение рацемического диоламина
 |
 3. Расщепление рацемата (+)- винной кислотой и получение левомицетина
3. Расщепление рацемата (+)- винной кислотой и получение левомицетина
ИдентификацияПрим. спектрометрические (ИК, УФ), хроматографические, хим. методы, а также различные физ-химические характеристики данных веществ (Тпл, уд. вращение). Для идентификации хлорамфеникола и его натриевой соли и сукцината хлорамфеникола используется ТСХ: неподв. фаза
– силикагель; подв. – смесь воды, метанола и хлороформа /для хлорамфеникола/ или ледяной уксусной кислоты, метанола и хлороформа
/для натриевой соли сукцината/. Проявляют хроматограммы под УФ-лампой. Для идентификации пальмитата хлорамфенкола используется обращённо- фазовая ТСХ: неподв. фаза - силанизированный силикагель, подв. – смесь этанола и водного раствора ацетата аммония. Хроматограмму проявляют р- ром дихлорфлуоресцеина и родамина в этиловом спирте. Химические реакции: 1)нагревание с NaOH;2) обнаружение хлорид-ионов в растворе после гидролиза;3) вз-вие с пиридином и щелочью;4) восстановление до аминогруппы, диазотирование и азосочетание;5) с цинком, бензоилхлоридом и хлоридом железа (III);6) частные реакции (гидролиз стеарата левомицетина, обнаружение ионов натрия в натриевой соли сукцината).
Испытания на чистотуПри контроле чистоты хлорамфеникола и его сложных эфиров определяют их физ. константы (удельное вращение, Тпл), кислотность
и щёлочность растворов, примеси хлоридов, потерю в массе при высушивании, сульфатную золу, стерильность, пирогенность. Хлорамфеникол устойчив в кристаллическом состоянии, но в водном растворе может подвергаться гидролизу. В лекарственных формах хлорамфеникола (капсулы, капли, глазная мазь) определяется примесь продукта гидролиза данного вещества – 2-амино- 1-(4-нитрофенил)пропандиола-1,3. Определение проводят методом ВЭЖХ (С18, подв. фаза – смесь водного раствора пентансульфоната натрия, ацетонитрила и ледяной уксусной кислоты 85:15:1, 272 нм).
ВP и USP определяют содержание в образцах пальмитата хлорамфеникола свободного хлорамфеникола (растворяют в ксилоле, экстрагируют водой, реэкстрагируют толуолом и далее измеряют оптическую плотность водного слоя, она не должна превышать определённой величины). Согласно BP в образце пальмитата хлорамфеникола определяются примеси родственных соединений (изомерного сложного эфира и дипальмитата). Определение проводится методом ТСХ (силикагель; метанол - хлороформ – циклогексан 10:40:50). В образце натриевой соли сукцината хлорамфеникола согласно BP определяются примеси хлорамфеникола и дисукцината. Определение проводится методом ВЭЖХ (С18, смесь водного раствора фосфорной кислоты и метанола). Согласно USP методом ВЭЖХ определяют примесь хлорамфеникола (С18, смесь аммиачно-фосфатного буфера и метанола).
Количественное определениеДля количественного определения хлорамфеникола и его сложных эфиров в субстанции и лек. формах используют: титриметрические методы (нитритометрия (после восстановления); аргентометрия или меркуриметрия (после гидролиза); иодометрическое (обратное, или титрование заместителя по реакции с CuSO4) цериметрическое куприметрическое (индикатор – мурексид, fэкв=2) спектрометрические (УФ-спектрофотометрия (водный раствор, 278 нм)); хроматографические методы (ВЭЖХ. Напр, по USP для хлорамфеникола, пальмитата хлорамфеникола в субстанции и лек. формах: С18, подв. фаза – смесь воды, метанола и ледяной уксусной кислоты, детектирование – фотометрическое (280 нм)).
211.
 |  |
Способы получения и методы контроля качества сульфаниламидов и триметоприма.
Исходное вещество для синтеза анилин. Сначала в анилине защищают аминогруппу, переводя её в карбаматную. Затем действием хлорсульфоновой кислоты получают сульфонилхлорид, который обрабатывают аммиаком или амином. В полученном соединении гидролизом (кислотным или щелочным) освобождают ароматическую аминогруппу.



 Триметопримбелый или желтовато белый порошок, очень мало растворим в воде, мало − в этаноле. ПодлинностьУФ-спектр (0,1М NaOH; максимум 287 нм и плечо 240- 250 нм), ИО + H2SO4 + щелочной р-р КМnO4, нагревают до кипения, затем + H2SO4 и НСНО. Охл., фильтруют. К фильтрату + СН2Сl2 − орг.
Триметопримбелый или желтовато белый порошок, очень мало растворим в воде, мало − в этаноле. ПодлинностьУФ-спектр (0,1М NaOH; максимум 287 нм и плечо 240- 250 нм), ИО + H2SO4 + щелочной р-р КМnO4, нагревают до кипения, затем + H2SO4 и НСНО. Охл., фильтруют. К фильтрату + СН2Сl2 − орг.
слой имеет зелёную флуоресц. Количественное определениеацидиметрия (безв. АсОН; 0,1 М HClO4; потенц. КТТ; 1:1). В таблетках − экстракция хлороформом из щел.среды, реэкстракция 1М АсОН и СФМ при 271 нм.
Сульфаниламиды− белые или белые с желтоватым оттенком кристалл. в-ва без запаха. Мало растворимы или практически нерастворимы в воде, этаноле, эфире, хлороформе. В ацетоне некоторые растворимы (сульфаниламид, сульфамет). Натриевые соли легко растворимы в воде и метаноле. Растворимы в кислотах и растворах щелочей (амфотерные свойства). Подлинность: ИК-спектры, ТСХ, реакция образования азокрасителя, в щелочной среде образуют продукты конденсации с 2,4-динитрохлорбензолом (желтого цвета) и в кислой среде окрашенные продукты конденсации с альдегидами типа шиффовых оснований, при термическом разложении плавы приобретают различную окраску. Испытаниена чистоту (отсутствие или предельное содержание органических примесей, сульфатов, хлоридов, сульфатной золы, тяжелых металлов, кислотность или щелочность, прозрачность, цветность). Определяют потерю в массе при высушивании. Для испытания на посторонние органические примеси используют ТСХ. Количественное определение: 1. нитритометрия(титрант 0,1 М р-р нитрита натрия). КТТ: с помощью внутренних индикаторов (тропеолин 00, нейтральный красный); внешних индикаторов (йодкрахмальная бумага) или потенциометрически. 2. СФМ(растворители 0,1 М растворы NaOH и HCl, УФ- спектры в области 210-360 нм). Сульфасалазин − 359 нм (растворяют в растворе
NaOH, затем + АсОН). 3. нейтрализацияв водно-ацетоновом растворе или в этаноле (индикатор тимолфталеин), в неводных растворителях (диметилформамид (индикатор тимоловый синий), в спирто-ацетоновой среде (индикатор метиловый оранжевый)).
212. Способы получения и методы контроля качества нитрофуранов.Исходным веществом для синтеза нитрофуранов - фурфурол, получаемый из пентозансодержащего сырья (кукурузные початки, овсяная и рисовая шелуха и т.д.) действием разбавленных минеральных кислот
(C5H8O4)n+ nH2O→ nC5H10O→ ФУРФУРОЛ
При нитровании фурфурола в мягких условиях (ацидофобность!) получают 5-нитрофурфурол:
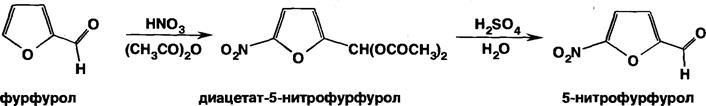
 |
Дальнейший синтез основан на конденсации 5-нитрофурфурола с различными веществами, содержащими аминогруппу по схеме:
| Свойства | Нитрофурал | Нитрофурантоин | Фуразолидо н | Нифуроксазид |
| Описани е | Жёлтый или коричневато -Ж. КП | ЖКП либо-жёлтый кристаллы кристаллы, без запаха либо почти без запаха | ЖКП | Ярко-желтый КП |
| Раствори мость | Вода–ПрН, , EtOH- М | Вода–ОМ, EtOH- ОМ | Вода–ОМ, EtOH-ОМ, эфир-ПрН | Вода–ПрН, EtOH - М, |
| Идентиф | ИК, УФ, ТСХ, | ИК, УФ, + ДМФА, + KOH (спирт.) –фиолетово- красное | ИК, + ДМФА, | ИК |
| икация | + ДМФА, + | + KOH | ||
| KOH (спирт.) | (спирт.) | |||
| –фиолетово- | –фиолетово- | |||
| красное | красное | |||
| Чистота | К или Щ, | СПр–ТСХ; ∆m, СЗ, | К или Щ, рН, | ТМ,СПр–ВЭЖХ; |
| рН,СПр–ВЭЖ | ОКОР, МЧ | ∆m, СЗ, | ∆m, СЗ, УПП при | |
| Х; ∆m, СЗ, | ОКОР, МЧ, | 367нм. Примесь | ||
| ОКОР, МЧ | Tпл 257- | п- | ||
| 259°С, | гидроксибензо- | |||
| Нитрофурал | гидразида | |||
| я диацетат - | –фотометрия с | |||
| ТСХ | фосф.-вольф.к | |||
| Кол | Спектрофото | Спектрофотометр ия (ДМФА + ацетатный БР; 367нм; УПП = 375).Таблетки – аналогично | Спектрофото | Алкалиметрия |
| опред | метрия | метрия | (ДМФА + вода; | |
| (ДМФА + | (ДМФА + | 0,1 МNaOH; | ||
| вода, 375нм, | вода, 367нм, | потенц. КТТ; | ||
| по | УПП = 750). | 1:1). | ||
| сравнению с | ||||
| ФСО) |
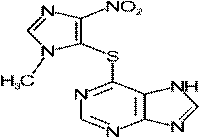
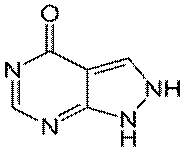
213. Способы получения и методы контроля качества нитроимидазолов.
Дата добавления: 2021-03-18; просмотров: 697;
Поиск по сайту

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
