СОЗДАНИЕ И АНАЛИЗ КЛОНОТЕК ГЕНОМОВ
Генетическая инженерия как основа современной биотехнологии зародилась в семидесятые годы ХХ ст. в недрах молекулярной биологии. К этому времени были расшифрованы механизмы основных матричных процессов, происходящие в клетках прокариот — репликация, транскрипция и трансляция, кроме того, эти процессы были воспроизведены in vitro. Была определена структура генетического кода и синтезирован первый ген (аланиновой тРНК дрожжей). Удалось выделить и изучить свойства некоторых ферментов, использующих в качестве субстрата ДНК (рестриктазы, лигазы, ДНК-полимеразы). Эмбриологи освоили методологию пересадки ядер соматических клеток животных (лягушки) взамен удаленного гаплоидного ядра, что позволило воспроизводить животных неполовым путем, т. е. клонировать (получать генетически идентичные особи).
Так, впервые появилась надежда на возможность замены определенных генов (например, дефектных) в зародышевых клетках на другие—полноценные, иными словами, осуществления генной терапии. Однако для этого необходимо было освоить методологию выделения генов, их идентификации, тиражирования, устойчивого культивирования в составе автономно реплицирующихся молекул, а также определения свойств продуктов этих генов. Все эти вопросы решает генетическая инженерия, под которой подразумевают комплекс молекулярно-генетических методов, позволяющих осуществить целенаправленное конструирование организмов путем манипуляций с их наследственным аппаратом.
Поскольку микроорганизмы, в частности бактерии, как одни из наиболее просто организованных форм живого, изучены гораздо лучше с генетической точки зрения, чем макроорганизмы, первые эксперименты по получению рекомбинантных ДНК были осуществлены именно с бактериальными клетками. И вскоре обнаружилась еще одна важная перспектива использования достижений генетической инженерии, а именно конструирование высокопродуктивных штаммов микроорганизмов—продуцентов биологически активных веществ, обладающих комплексом заданных свойств.
В данной главе охарактеризованы основные методы, позволяющие получать определенные гены, вводить их в состав векторных молекул для клонирования в клетках-реципиентах, идентифицировать гены в клонотеках, определять последовательность нуклеотидов в ДНК. Знание данной методологии необходимо для современных биотехнологов, использующих для создания организмов — продуцентов нужных веществ достижения генетической инженерии.
Для конструирования организмов с заданными свойствами требуется иметь набор генов, детерминирующих желаемые функции (утилизация определенных субстратов, биосинтез и секреция определенных продуктов, устойчивость к определенным физическим и химическим факторам, деградация ксенобиотиков и т. п.). Такие гены можно выделить из любых организмов, обладающих соответствующими свойствами, но для этого вначале следует получить клонотеку генома данного организма и охарактеризовать ее. Под клонотекой генома понимают набор клонов бактерий или бактериофагов, каждый из которых содержит один тип рекомбинантной ДНК, а в совокупности — весь геном изучаемого организма, фрагменты которого распределены по отдельным клонам.
Получение генов
Используется три основных способа получения нужных генов: выделение из состава ДНК, химико-ферментативный синтез (in vitro), транскрипция изолированной из клетки матричной РНК с помощью обратной транскриптазы (РНК-зависимой ДНК-полимеразы, ревертазы).
Выделение генов из ДНК. Большинство методик в генетической инженерии основано на вырезании из молекул ДНК определенных фрагментов и соединении их с другими фрагментами для получения рекомбинантных (химерных) ДНК. При этом фрагментация выделенной из клеток ДНК осуществляется с помощью ферментов, которые способны расщеплять ДНК в строго определенных сайтах. В качестве таких ферментов используются эндонуклеазы рестрикции (рестриктазы), общая характеристика которых дана в главе 2. В генетической инженерии используются рестриктазы, образующие в ДНК однонитевые (липкие) концы (при разрезе уступом), а также те, что формируют в ДНК двухнитевые (тупые) концы (при расщеплении по середине узнаваемого участка нуклеотидных пар). Примером рестриктазы первого типа, образующей липкие концы, является Eco R1 (рис. 2.1), а рестриктазой второго типа является, например, Hind II.
Образование липких концов при расщеплении ДНК имеет преимущество, которое состоит в возможности реассоциации образованных фрагментов по гомополимерным липким концам (содержат комплементарные нуклеотиды). В таком случае появляется возможность формирования ассоциатов из фрагментов, принадлежащих разным молекулам ДНК, что и лежит в основе большинства генноинженерных манипуляций по получению рекомбинантных ДНК (рис. 20.1). Образующиеся спонтанно ассоциаты могут быть превращены в целые молекулы путем «сшивания» с помощью ДНК-лигаз.
Следует указать, что в обычных условиях липкие концы в составе одной молекулы могут удерживаться друг относительно друга с помощью водородных связей между комплементарными основаниями. Однако

комплементарные цепочки легко разделить при небольшом нагревании растворов ДНК (денатурация ДНК). При охлаждении липкие концы гибридизуются вновь за счет восстановления водородных связей при соблюдении принципа комплементарности (отжиг). В результате отжига набора фрагментов, полученных при воздействии на разные молекулы ДНК одной и той же рестриктазой, могут образоваться как исходные молекулы ДНК, так и их гибриды—рекомбинантные ДНК.
В 1972 г. в Стэнфорде Дж. Мерц и Р. Дэвис осуществили первый подобный эксперимент. Позднее оказалось, что не все геномы могут содержать сайты рестрикции для используемых рестриктаз. Особенно это касается небольших молекул: плазмид и профагов, которые настолько малы, что содержат лишь по несколько сайтов рестрикции для небольшого числа рестриктаз. Данное обстоятельство существенно ограничивает возможности метода, поэтому была разработана методология введения в геном фрагментов ДНК, содержащих необходимые сайты рестрикции.
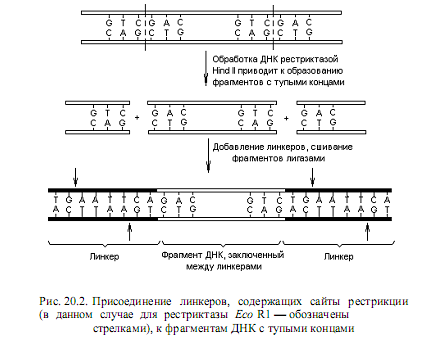
Для этого используют искусственно синтезированные олигонуклеотидные ДНК с тупыми концами (линкеры). Линкеры синтезируют таким образом, чтобы в их составе содержались известные сайты рестрикции (предварительно требуется установить последовательность нуклеотидов в этих сайтах). ДНК для введения линкеров в ее состав расщепляют одной из рестриктаз, образующих тупые концы, а затем «сшивают» полученные фрагменты с линкерами с помощью лигаз (рис. 20.2). Альтернативным методом получения тупых концов является механический разрыв крупных молекул ДНК при быстром перемешивании раствора или продавливании его через узкое отверстие. В результате этих манипуляций фрагменты ДНК приобретают сайты рестрикции внутри добавочных последовательностей (линкеров).
Теперь полученный фрагмент ДНК, содержащий требуемые сайты рестрикции, можно присоединить к другой молекуле ДНК (например, к вектору), обработанной такой же рестриктазой, либо превратить в кольцевую форму путем сшивания взаимно комплементарных концов.
Описанные методы выделения генов в составе фрагментов ДНК с помощью рестрикционных ферментов широко распространены, но имеют ряд недостатков. Во-первых, не всегда удается подобрать рестриктазы, позволяющие вырезать из ДНК именно тот участок, в котором содер-
жится интересующий ген. Во-вторых, в составе вырезанного фрагмента ДНК могут оказаться затрудняющие дальнейшее использование гена последовательности, например интроны в эукариотических генах. В данном случае рекомбинантные ДНК не смогут экспрессироваться в прокариотических клетках, поскольку последние не обладают способностью к сплайсингу (глава3).
Химико-ферментативный синтез генов. С помощью этого метода синтезированы, а впоследствии клонированы гены, определяющие структуру таких гормонов, как инсулин и соматостатин, а также лейкоцитарный интерферон человека. Синтез гена интерферона осуществлен в СССР в 1984 г. под руководством академика М.Н. Колосова.
Суть метода сводится к следующему: in vitro осуществляют химический синтез коротких (8— 16 нуклеотидов) одноцепочечных фрагментов ДНК, которые затем соединяют с помощью лигаз и отжигают (дают возможность образоваться двухнитевым молекулам ДНК). Для этого метода необходимо знать последовательность нуклеотидов в гене, поскольку синтез осуществляется без матрицы. Ее устанавливают обычно исходя из аминокислотной последовательности соответствующего полипептида, однако из-за вырожденности генетического кода определить истинную нуклеотидную последовательность гена оказывается невозможным. Установить истинную структуру гена можно методом секвенирования ДНК, однако для этого требуется выделить и клонировать соответствующий ген.
Этап химического синтеза олигонуклеотидов в настоящее время полностью автоматизирован. Метод основан на специфической защите 5’ или 3’-конца моно- или олигонуклеотида, предотвращающей его вступление в химические реакции. При необходимости блокирующие группы, с помощью которых осуществляют модификацию, можно удалить обработкой кислотой либо щелочью. Цикл химического синтеза ДНК включает конденсацию нуклеотидов, удаление той или иной блокирующей группы и дальнейшую конденсацию. Модификацией метода является прикрепление первого нуклеотида к твердому носителю и добавление следующих нуклеотидов по одному после промывки носителя на каждом таком этапе.
Воссоединение одноцепочечных фрагментов с помощью лигаз требует фосфорилирования 5’-концов, что осуществляется с использованием фермента полинуклеотидкиназы и АТР. Одноцепочечные ДНК можно превратить в двухцепочечные либо в процессе отжига с комплементарной антипараллельной цепью, также синтезированной химическим путем, либо при достройке комплементарной цепи ферментом (обычно используют ДНК-полимеразу I). При сочетании химического синтеза и ферментативных этапов, например, из 66 коротких синтетических фрагментов был воссоздан ген инсулина длиной 514 п. н.
Ферментативный синтез генов. Возможны два принципиально различающихся способа ферментативного синтеза ДНК. Один из них не требует присутствия матрицы и осуществляется по программе, задаваемой экспериментатором. Такой синтез катализирует бактериальный фермент полинуклеотидфосфорилаза, специфичный в отношении рибонуклеотидов, но способный с меньшей скоростью и к полимеризации цепей ДНК. Для такого синтеза необходим праймер, включающий не менее 3 нуклеотидов. Реакции полимеризации имеют некоторые ограничения и с трудом поддаются контролю.
Другой способ ферментативного синтеза ДНК предполагает участие матрицы, которой на первом этапе служит мРНК, выделенная из клетки. Практически все мРНК эукариот имеют на 3ў-конце «шлейф» (полиаденилатную последовательность). Этот участок используют для образования затравки для комплементарной цепи ДНК: к мРНК добавляют короткие последовательности, состоящие из тимидилатов, которые в результате отжига гибридизуются с полиаденилатами (рис. 20.3). Обратная транскриптаза в присутствии набора дезоксирибонуклеотидов катализирует их присоединение к затравке в последовательности, определяемой мРНК, в результате чего образуется двухнитевый гибрид РНК-ДНК. По невыясненным пока причинам новосинтезированная цепь ДНК имеет на конце шпильку (рис.20.3), которая возникает только при реакции in vitro (по-видимому, из-за того, что фермент «поворачивает вспять»). Эту шпильку используют в качестве затравки для синтеза вто-

рой цепи ДНК. На следующем этапе осуществляют деградацию РНК с помощью рибонуклеаз либо в ходе щелочного гидролиза, а оставшуюся однонитевую ДНК со шпилькой используют как матрицу для синтеза второй цепи ДНК (ДНК-полимераза 1). На заключительном этапе шпильку расщепляют с помощью нуклеазы S1, которая специфически гидролизует одноцепочечные участки нуклеиновых кислот. Так образуется двухцепочечная «комплементарная» ДНК, или кДНК (в названии отражено основное ее отличие—комплементарность мРНК).
Существуют модификации описанного метода, позволяющие избежать многих его недостатков, в частности синтеза неполных копий РНК (особенно в случае длинных мРНК). Одной из разновидностей метода является синтез кДНК непосредственно на векторе.
Дата добавления: 2016-05-30; просмотров: 2768;
Поиск по сайту
Узнать еще
- Case-study (анализ конкретных ситуаций, ситуационный анализ)
- II Расчет и анализ трехфазных цепей
- II. Зарождение национального самосознания. Реформационное движение, создание Индийского национального конгресса
- II. Качественный контроль (социологический анализ).
- III и IV нейроны слухового пути. Третьи и четвертые нейроны слухового проводящего пути. Ядра слухового анализатора. Признаки поражения слухового пути.
- VII. Анализ характера
- І. Анализаторы І сигнальной системы
- ІІ. Анализаторы ІІ сигнальной системы

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
