Електрохімічно діючі середовища
Дуже поширеними є робочі середовища, що внаслідок адсорбційної дії на фізико-механічні властивості металу викликають корозію, причому в рідких середовищах і при найбільш поширених робочих температурах цей вплив має переважно електрохімічний характер.
Найбільш поширеним видом електрохімічно діючих середовищ є вологе повітря, в якому експлуатується близько 80% усіх металевих конструкцій. Далі за ступенем розповсюдження іде вода і водні розчини, особливо такі природні середовища, як грунтова і морська вода. Велика кількість деталей машин та механізмів, зокрема деталі насосів, гідротурбін, морських споруд тощо, експлуатуються саме в цих середовищах. Деталі хімічної апаратури працюють в ще більш корозійно-агресивних середовищах, включаючи кислоти, луги і солі.
Електрохімічна корозія відбувається у водяних розчинах солей, кислот і лугів, в чистій воді і розплавах солей.
Всі ці середовища, будучи електролітами, характеризуються присутністю іонів і поділяються на дві групи — слабкі і сильні. До слабких належить більшість органічних кислот та лугів, до сильних — всі солі і більшість неорганічних кислот. Розчини слабких електролітів дисоційовані на іони лише частково, причому ступінь дисоціації знижується з концентрацією, сильні ж електроліти повністю дисоційовані на іони. Зменшення електропровідності сильних електролітів із збільшенням концентрації, вище від граничного значення, пояснюється наявністю електростатичних сил взаємодії між протилежно зарядженими іонами.
У відповідності з існуючими уявленнями, поверхня сталі в електроліті являє собою короткозамкнений багато електродний гальванічний елемент, що складається :3 великої кількості мікроелементів, і його виникнення зв’язане, по-перше, з неоднорідністю металевої фази, яка змінюється внаслідок термічної і механічної обробки, по-друге, з неоднорідністю плівок окисів і забруднень на поверхні металу, і, по-третє, з неоднорідністю рідкої фази, що обумовлена різністю концентрацій як власних іонів металу в електроліті, так і іонів розчиненої речовини та газів.
Електрохімічна неоднорідність поверхні сталі, що виникає в результаті згаданих вище причин, викликає появу на поверхні сталі катодних і анодних ділянок металу. Як відомо, ті ділянки металу, які мають більш негативний електродний потенціал, утворюють аноди, ділянки з більш позитивним електродним потенціалом— катоди. Матеріальний ефект втрат металу від електрохімічної корозії зосереджується на анодних ділянках.
Під електродним потенціалом металу при зануренні його в електроліт розуміють різницю потенціалів між поверхнею металу і електролітом. Ця різниця викликана взаємодією металу з електролітом: переходом іонів металу в електроліт і віддачею металом електронів на анодних ділянках (окислювальний процес) та асиміляцією надлишкових електронів іонами (атомами, молекулами), що перебувають в електроліті на катодних ділянках (відновлювальний процес).
У зв’язку з відсутністю безпосереднього експериментального визначення різниці потенціалів між двома фазам — електродом і електролітом (абсолютної величини електродних потенціалів) — визначають величину с. р. с. між досліджуваним електродом і яким-небудь одним (однаковим для усіх випадків) електродом, потенціал якого умовно прийнятий за нуль (стандартний водневий електрод) або потенціал якого вже відомий (електрод порівняння). Визначена таким чином е. р. с. характеризуватиме потенціал електрода, що нас цікавить, відносно деякого умовного нуля.
Загальний електродний потенціал поверхні металу значною мірою характеризує його термодинамічну стійкість проти корозійного руйнування: прийнято вважати, що більш негативне значення потенціалу відповідає менш стійкому стану металу, а облагороджування потенціалу (зсув до більш позитивних значень) вказує на підвищення стійкості проти корозії, хоч таке твердження не можна вважати універсальним та однозначним. Безсумнівно, що при утворенні корозійних пар з ділянок поверхні з різною механічною обробкою, які відрізняються рівнем власного стаціонарного потенціалу, анодом буде найбільш електронегативна ділянка і зона інтенсивно кородуватиме, в той час як катодна ділянка буде, навпаки, захищатися від корозії. Тому величини загальних і локальних електродних потенціалів є необхідними енергетичними характеристиками електрохімічного стану поверхні.
Величина електродних потенціалів визначається переважно компенсаційним методом. Він полягає в тому, що вимірювана е. р. с. компенсується відомою напругою якого-небудь постійного джерела струму. Коли замкнути електроди на вимірювальний прилад, який працює лише при протіканні електричного струму, то знайдена величина е. р. с. буде меншою, ніж величина, визначена як різниця рівноважних потенціалів досліджуваного і порівняльного електродів при розімкненому колі (через поляризацію, зміну концентрації розчину біля поверхні електродів та ін.). Компенсаційний метод дозволяє проводити вимірювання е. р. с. без протікання струму в досліджуваному гальванічному елементі.
Корозійно-агресивне середовище викликає розчинення всіх доступних для нього анодних ділянок поверхні металу. На цих ділянках іон атома металу переходить в розчин, причому звільняються електрони; засвоєння цих вільних електронів елементами розчину відбувається на катодних ділянках поверхні поділу метал-електроліт. При замиканні в електроліті двох ділянок металу з різними потенціалами відбувається перехід електронів з ділянок з вищим потенціалом на ділянки з нижчим потенціалом. Цей перехід електронів вирівнює значення потенціалів електродів, що приводить до повної поляризації всього металу, але переважно повної поляризації нема із-за анодного і катодного процесів. При анодному процесі біля анодних ділянок металу і гідратованих іонів металу в електроліті виникають некомпенсовані електрони; при катодному — відбувається асиміляція електронів деякими іонами або молекулами розчину, що є деполяризаторами і забезпечують протікання електрохімічного процесу. Деполяризація може протікати за рахунок іонів, нейтральних молекул розчину і нерозчинних плівок.
В кислих середовищах зменшення поляризації катода в основному відбувається внаслідок високої концентрації водню (водневої деполяризації). В нейтральних середовищах спостерігається в основному киснева деполяризація за рахунок кисню, розчиненого в середовищі, а в лужних середовищах великого значення в процесі деполяризації набувають захисні плівки окисів. Слід, проте, відзначити, що корозія заліза термодинамічно можлива (в нейтральних середовищах. Наприклад, при t = 25° С, рН 7 для водневої поляризації потрібно, щоб потенціал металу був більш негативним, ніж — 0,228 в, що можливе.
В корозійних процесах з водневою деполяризацією електрони звільняються при іонізації металу на анодних ділянках і витрачаються на розряд іонів водню на катодних ділянках. Корозійний потенціал в цьому випадку встановлюється при еквівалентній кількості металу, який переходить в розчин, і водню, що виділяється при цьому. Такий хід процесу не обумовлюється просторовим розподілом катодного та анодного процесів. Із збільшенням рН середовища значення корозійного потенціалу стає все більш позитивним. Корозія з водневою деполяризацією вже не може проходити, якщо рівноважний потенціал металу більш негативний, ніж рівноважний водневий потенціал.
В процесах з кисневою деполяризацією електрони, що звільняються при іонізації металу, йдуть на іонізацію розчиненого кисню. Швидкість процесу часто визначається швидкістю дифузії кисню до металу і зростає з підвищенням концентрації кисню в розчині. Між процесами з чисто водневою і чисто кисневою деполяризацією існує ряд перехідних ступенів.
Із сказаного видно, що електрохімічна корозія сталі залежить від природи електроліту і по-різному проходить в кислих, лужних та нейтральних розчинах. Зміна рН розчину по-різному впливає на корозійні процеси різних металів, у зв’язку з чим для різних металів є певні значення рН для оптимальних корозійних процесів. На анодний процес переходу іонів металу в розчин зміна рН безпосередньо не впливає; рН впливає лише на процеси, які мають місце на катодних ділянках металу. При зменшенні рН розчину в кислих середовищах збільшується концентрація іонів водню, що полегшує їх розрядження. Внаслідок цього підвищується швидкість корозії. Таке явище можливе, звичайно, лише у випадку, коли з продуктів корозії не виникають плівки, що пасивують метал від впливу середовища. Якщо такі плівки утворюються, то швидкість корозійного (рис. 5.2) процесу значно зменшується і корозія може навіть припинитися.
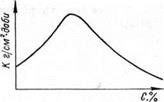
Рисунок 5.2 - Залежність швидкості корозії К від концентрації С нейтральної солі в розчині
Швидкість корозії при високих рН (в розчинах лугів) характеризується розчинністю продуктів корозії. Якщо гідрати алюмінію, цинку та свинцю в їдких лугах досить легко розчиняються і метал втрачає плівку, що призводить до різкого підвищення швидкості корозії, то залізо, нікель, кадмій і магній в середовищах з високим рН не дають розчинних комплексних сполук, у зв’язку з чим стають більш корозійно стійкими. Внаслідок цього корозія сталі з підвищенням рН зменшується і при рН 13 швидкість корозії практично дорівнює нулеві, незалежно від кількості розчиненого кисню в рідкому середовищі. Проте при високих температурах і високих концентраціях лугів корозія сталі активується за рахунок виникнення розчинних комплексних сполук (фератів). При високих температурах в лужних розчинах спостерігається зміщення електродних потенціалів сталі в бік катоду зі збільшенням концентрації, наприклад, їдкого натрію. Тому підвищення рН лужних середовищ зменшує можливість анодного розчинення металу і збільшує можливість наводнювання сталі, яке, на думку деяких авторів, є першопричиною лужної крихкості.
Домішки до лужних середовищ, які зміщують електродний потенціал в бік анода (наприклад, NaNO3) знищують можливість розвитку процесів, зв’язаних з утворенням водню, тобто зменшують лужну крихкість, тоді як домішки, що зміщують електродний потенціал в бік катода, навпаки, підвищують можливість утворення водню, тобто збільшують лужну крихкість. До таких домішок належить Na2SiO3.
Вплив кисню на швидкість корозії сталі двоякий: з одного боку, кисень як деполяризатор збільшує швидкість катодного процесу, а з другого, —• підвищує стабільність захисних плівок, що гальмує корозійний процес. Мала і середня концентрація кисню в розчині збільшує швидкість корозії. З ростом рН розчину дія кисню виявляється більше.
Аналогічно кисню на корозійний процес сталі діють, в залежності від їх кількості, як сповільнювачі (інгібітори) або прискорювачі (стимулятори) корозії бікарбонат натрію (NаНСО3), кислий фосфат натрію (NaPO4) і хромати лужних металів (К2Сг07; Na2Cr2O7). Крім перелічених анодних сповільнювачів, існують ще катодні сповільнювачі — Са(НСОз)2; ZnSO4, деякі сполуки нікелю, магнію; колоїдні сповільнювачі — желатин, агар- агар та ін.
Швидкість корозії більшості металів в розчинах солей та кислот залежить від характеру іона. В результаті гідролізу вони можуть знижувати або підвищувати рн розчину. Залежність швидкості корозії К від концентрації С нейтральних солей в нерухомій системі ілюструє рис. 5.2. Аналогічний вигляд мають криві для кислот. Швидкість корозії в соляних розчинах залежить від характеру аніонів та катіонів середовища, розчинності продуктів корозії, стійкості утвореної на поверхні металу захисної плівки та її адгезії. Наприклад, для сталей розчини солей NН4С1; NaС1; К2S04, що утворюють з залізом розчинні сполуки, посилюють корозію, а такі солі, як Na2CO3; Кз[Fе(СN)6]; К4[Fе(СN)6], дають важкорозчинні сполуки, внаслідок чого зменшують корозію.
Швидкість корозійного процесу металу із збільшенням концентрації солі або кислоти зростає внаслідок підвищення електропровідності розчину та дії аніонів, таких як СІ; S04, що зменшують захисні властивості плівок. Проте посилення концентрації нейтральних солей зменшує розчинність кисню, що викликає зниження швидкості корозії. Розчинність кисню залежить від характеру солі, концентрації її в розчині і температури.
Хлор є активатором анодного процесу. Для більшості металів хлор, як і інші галоїди (йод і бром), внаслідок дуже малих розмірів іона має здатність проникати в найменші пори і порушує окисні плівки на металах, що прискорює процес руйнування останніх. Крім цього, хлор здатний адсорбційно витісняти кисень з поверхні металу. Іони галоїдів, навіть при незначній концентрації в розчині, в результаті безперервного руху до кородуючої металевої поверхні можуть мати на ній високу щільність. Температура впливає і на характер корозії: до 80 °С сталь у водопровідній воді зазнає місцевої корозії, а вище цієї температури корозія набуває рівномірного характеру.
Вплив тиску на корозійні процеси зв’язаний з розчинністю в електроліті газів та з гідролізом розчинених в ньому солей. У відкритій системі з підвищенням тиску збільшується розчинність кисню і посилюється гідроліз. Обидва ці фактори прискорюють корозію. Швидкість корозії сталі у воді, що вміщує вуглекислоту, зростає при підвищенні тиску до 20 ат, а потім зменшується.
Гази, розчинені в електроліті (Н2; 02; С02; Н2S та ін.), беруть участь в електрохімічних процесах корозії, наявність їх в електроліті при збільшенні тиску викликає прискорення корозії сталі. Корозія сталі під впливом розчинених у воді 02, С02, Н2S в концентраціях, що зустрічаються при експлуатації нафтових свердловин, під дією розчиненого кисню майже пропорційна його концентрації в розчині, але лише до певної межі. Корозія в цьому випадку має крапчастий характер, типовий для кисневої корозії. В результаті корозії під впливом розчиненого вуглекислого газу щербин на поверхні металу було менше, ніж при корозії під дією кисню, але були вони глибшими.
При дослідженні корозії сталі в сірководневих розчинах спостерігалося різке її збільшення в межах концентрації Н2S від 0 до 150 мг/л з ростом концентрації від 150 до 400 мг/л швидкість корозії залишалася на сталому рівні, але потім різко знижувалась. Починаючи з концентрації сірководню 1600 мг/л, швидкість корозії знову набувала постійного значення.
Сталевіі зразки при корозії в сірководневій воді мали невелику кількість щербин, але значну кількість здyтин та розшарувань; поверхня їх була покрита темною, щільно прилягаючою до металу плівкою сульфідів заліза. Деякі дослідники вважають, що сульфіди заліза відіграють роль сповільнювача корозії на початкових стадіях корозійного процесу сталі, а на більш пізніх стадіях, навпаки, стають активатором цього процесу. Існує пояснення пасивності сталі при взаємодії з концентрованими розчинами Н2S, згідно з яким хімічний склад плівки різний при різних концентраціях Н2S. Вважають також, що при високих концентраціях можливе утворення напівсульфідів.
Дослідження кінетики сірководневої корозії показало, що сірководень, адсорбуючись на поверхні металу, прискорює анодний процес, полегшуючи перехід іонів металу в розчин. Про роль сірководню в катодному процесі при корозії металів в слабокислих і нейтральних електролітах єдиної думки нема. Одні автори вважають, що сірководень прискорює, а інші, навпаки, що він сповільнює катодний процес.
В результаті дослідження була виявлена невідома раніше структура сульфіду заліза, утвореного на сталі в початковий період кородування у вигляді тонкої плівки. Цей сульфід, названий «кенсайт», має формулу Fe9S8 і характеризується кубічною кристалічною граткою. Виявилося, що в початковий період ця плівка захищає сталь від корозії, згодом при потовщенні плівки процес корозії прискорюється. На наступній стадії корозії м’якої сталі в водних розчинах Н2S утворюються плівки піротину (Fe0,875S) і піріту (FeS) , які наростають на початковій плівці з Редвд і знову ж таки захищають метал від корозії. В присутності С02 або вільної S і при пропусканні Н2S або суміші Н2S + С02 через соляний розчин характер продуктів корозії змінюється: сульфід Fe9S8 існує лише у вигляді товстої облипаючої плівки, що не захищає сталь.
Властивості середовища, які обумовлюють наводнювання при корозії або травленні сталі, визначаються концентрацією водневих іонів в розчині і концентрацією розчину. Із зменшенням рН розчину кислих середовищ полегшується розряд іонів водню на поверхні металу і посилюється його наводнювання. Слід відзначити, що інтенсивність наводнювання різна при однакових величинах рН для різних кислот. Наприклад, наводнювання спостерігається в розчині фенолу вже при рН<7, вуглецевої кислоти при рН<6, тоді як в розчинах НС1 і Н2S02 лише при рH4.
Концентрація розчинів кислот при травленні впливає на інтенсивність наводнювання. Діапазон концентрацій, що викликають інтєнсиенє наводнювання, більший у сильно іонізованих кислот (зокрема, сірчаної і соляної) і менший у кислот середньої сили (наприклад, оцтової і фосфорної). Найбільш інтенсивне наводнювання і викликана ним зміна механічних властивостей металу спостерігаються при травленні в сірчаній кислоті; травлення ж в соляній кислоті дає менший ефект при тих же концентраціях і режимах травлення. Характерно, що вплив концентрації на інтенсивність наводнювання прямо протилежний для розчинів Н2S04 і НС1; якщо для розчину сірчаної кислоти (у всякому разі при збільшенні концентрації розчину до 10 н.) має місце лінійне збільшення швидкості дифузії і кількості оклюдованого сталлю водню, то для розчинів НС1 з ростом концентрації наводнювання сталі знижується. Це пояснюється різною дією іонів S04— і С1~ на розряд іонів водню. Найбільшої водневої крихкості маловуглецева сталь набуває при травленні в азотній кислоті невеликих концентрацій (0,3-0,8 н.).
Можна вважати, що наводнювання сталі прямо залежить від швидкості корозії з водневою деполяризацією, тому властивості середовища, які викликають посилення корозії, підсилюють і наводнювання.
Досить поширеним середовищем є морська вода. Хімічний склад різних морів не однаковий, але морська вода складається переважно з 3%-ного розчину NаСІ і деяких інших солей, причому в при поверхневому шарі вона сильно насичена киснем і продуктами життєдіяльності морських організмів. Корозія в морській воді є електрохімічною і відбувається з нерівномірною «диференційною» аерацією.
На корозійні процеси металу в морській воді великий вплив має обростання живими організмами — досить небажане явище для протікання корозії. Рослинні організми моря створюють навколо себе слаболужне середовище, тваринні — слабокисле, залежно від пори року і сонячного опромінення, що зумовлює вид корозії.
У морській воді глибин морів та океанів крім звичайних компонентів є ще значна кількість розчинених газів, шкідливих для фізико-механічних властивостей металів, таких як сірководень, що треба мати на увазі при розрахунку на міцність глибоко водневих апаратів та споруд.
Спостерігається також явище зневоднювання конструкційної сталі в розплаві NaС1. Деякі розплави вибірно вимивають із сталі легуючі елементи. Очевидно механізм корозії твердих металів в розплавах солей та лугів полягає в тому, що іони металу під дією силових полів аніонів розплаву переходять в розплав з анодних ділянок. Надлишок електронів в металі перетікає на катодні ділянки, де вони асоціюються деполяризаторами, якими є кисень і катіони розплаву, здатні при певних умовах на відновлення.
В умовах дії на твердий метал механічних напружень відбувається активація певних ділянок твердого металу з підсиленням названого вище процесу. Механічні напруження, крім того, руйнують захисні окисні плівки, чим посилюють процес електрохімічної корозії- Активація певних ділянок твердого металу сприяє також його розчиненню в розплаві солей, зокрема дифузії хрому по границях зерен в розплав, в результаті чого утворюються видимі пустоти і спостерігається між зеренне руйнування металу. В нітратно-нітритному розплаві в процесі деформації сталі відбувається активна дифузія азоту в напружений метал, що впливає на його механічні властивості.
Корозійні процеси, що протікають в металі, який піддають дії постійних або циклічних напружень, викликають або корозійне розтріскування металу, або його корозійну втому. Руйнування завжди відбувається по площинах дії найбільших напружень розтягу. В цьому випадку тріщини розміщуються під кутом 90° до силового потоку, а в зразках, що піддаються крученню, — під кутом 45° до площини зразка.
Корозійне розтріскування всіх металів, крім нержавіючої сталі і латуні, відбувається, як правило, міжкристалітно; це ж стосується і водневого розтріскування. Нержавіюча сталь і латунь можуть розтріскуватися як міжкристалітно, так і транскристалітно. Розвиток тріщин проходить окремими стрибками з характерним тріском, що вказує на механічне руйнування окремих ділянок металу і його окисів в тріщині, яка росте під впливом середовища й навантаження. Зародження тріщин та їх розвиток, очевидно, протікає під впливом електрохімічного та адсорбційного процесів і наводнювання катодних ділянок металу при корозії з водневою деполяризацією.
На корозійне розтріскування та втому впливають концентрація напружень розтягу або напруження від кручення. Деякі автори вважають, що існує граничне напруження, нижче якого ці явища не спостерігаються, але в той же час вказують, що напруження, які перевищують границю текучості, завжди небезпечні. Очевидно, безпечного напруження не існує, тому що крива корозійної втоми та тривалої міцності (крива корозійного розтріскування) весь час наближається до осі абсцис.
Однією з характерних і важливих в практичному відношенні особливостей високоміцних загартованих сталей є їх схильність до корозійного розтріскування та сповільненого руйнування, що може відбуватися під впливом тривалих зовнішніх навантажень та середовищ і навіть як результат дії лише внутрішніх напружень.
Схильність до сповільненого руйнування посилюється з ростом нерівноважності структурного стану мартенситу, в тому числі при різкому гартуванні з підвищених температур, збільшенні вмісту вуглецю в сталі, знижені температури (відпуску).
Як вже говорилося, при корозійній втомі і розтріскуванні може відбуватися транскристалітне, інтеркристалітне та змішане руйнування. Спостереження за розвитком розтріскування показали, що початку появи видимих тріщин передує деякий скритий інкубаційний період протікання корозійного розтріскування, тривалість якого неоднакова для різних металів і умов експерименту. Тому можна виділити три характерних періоди: інкубаційний (зародження тріщини); період розвитку корозійних тріщин; кінцеве спонтанне руйнування по одній або кількох найбільш розвинених тріщинах.
Інтенсивність утворення тріщин визначається в основному розвитком другої стадії процесу: його протікання обумовлене великими напруженнями розтягу, у місцях наявних концентраторів напружень й зароджуються тріщини (перший інкубаційний період). Очевидно, що в початковий період корозійного розтріскування переважний вплив на його розвиток має стан поверхні і наявність на ній концентраторів напружень. При цьому різниця у величині напружень розтягу на дні концентраторів і на поверхні деталі в міру розвитку початкових концентраторів буде зростати. В цьому випадку полегшується протікання анодного процесу на дні тріщини.
В такому ж напрямку діє так званий щілинний ефект, обумовлений утрудненим доступом кисню або інших деполяризаторів до дна тріщини, заповненої продуктами корозії, при одночасному хорошому надходженні його до відкритої поверхні деталі. Внаслідок цього збільшується різниця потенціалів між дном тріщини і сусідніми ділянками, в результаті чого швидкість корозії зростає. Розпирання тріщини продуктами корозії і утруднене надходження туди електроліту сприяють підвищенню катодної поляризації ділянок і переміщують катодну зону по стінках тріщини до зовнішньої поверхні. Разом з тим режим корозійного елемента може збільшувати ефективність роботи катодних ділянок, розміщених поблизу дна тріщини, тому що на відміну від поверхні на стінках тріщини відновлення захисних плівок майже неможливе.
Отже, при розвитку корозійних тріщин місце протікання корозійного катодного процесу визначається взаємодією таких двох діючих в протилежному напрямку факторів: зміщенням катодних ділянок до поверхні деталі внаслідок утруднення доступу деполяризатора в тріщину та інтенсифікацією катодного процесу поблизу анодних зон, викликаною руйнуванням захисних плівок.
Корозійне розтріскування та втому металу в більшості випадків пояснюють з позицій електрохімічної теорії корозії. Але сам електрохімічний процес не може пояснити всі явища та особливості, зв’язані з корозійним розтріскуванням та втомою. Якщо б електрохімічні процеси у всіх випадках були визначаючими, то будь-який електроліт з досить високою іонною провідністю повинен був би викликати корозійне розтріскування та втому, оскільки анодні і катодні процеси майже не змінюються при переході від одного електропровідного розчину до другого.
Найбільш повно дію корозійних середовищ та напружень на метал пояснює адсорбційно-електрохімічний механізм цього явища. Згідно з цією гіпотезою, перш ніж почнеться розчинення анодних ділянок під впливом корозійного середовища, на поверхні деталі має місце адсорбція поверхнево-активних елементів середовища — специфічна адсорбція іонів з електроліту, а у випадку корозії з водневою деполяризацією — адсорбція іонів водню на катодних ділянках поверхні, що викликає крихкість. Внаслідок адсорбції відбувається термодинамічно неминуче зниження поверхневої енергії, а разом з тим полегшення утворення тріщин. Далі починають протікати корозійні процеси, які призводять до росту тріщин. Розвитку корозійних тріщин сприяють розклинююча дія продуктів корозії та напруження розтягу. Кількість продуктів корозії з часом збільшується і залежить від агресивності середовища і стійкості металу. Напруження посилюють корозійний процес і викликають руйнування плівки окисів, яка може загальмувати і навіть повністю припинити корозію. В результаті розриву плівки окисів корозійний процес відбувається на не- захищених поверхнях деталей.
Таким чином, корозійне розтріскування та втома починаються процесом адсорбційного полегшення утворення субмікроскопічних тріщин під впливом зовнішніх напружень.
[3] Доцільніше будувати діаграму в координатах 5—<7, де — дійсне поперечне звуження,,границею якого є 100%.
[5] Приповерхневим шаром металу називається шар із зміненими фізико-хімічними властивостями, що обумовлене впливом обробки поверхні і способами її одержання.
[6] Адсорбція, зумовлена електростатичними силами для металів, як відомо [93], не залежить від ультрамікрошорсткості.
Дата добавления: 2016-06-29; просмотров: 1456;
Поиск по сайту
Узнать еще
- Іонізуюче випромінювання -це фізичні фактори зовнішнього середовища
- Аналіз зрізів внутрішнього середовища і внутрішніх ситуаційних змінних.
- Вимушенi коливання матерiальної точки без опору середовища. Резонанс
- Вплив корозійного середовища на швидкість корозії. Електрохімічний захист.
- ВПЛИВ СЕРЕДОВИЩА НА ВЛАСТИВОСТІ
- Лекція 3. Тема: Мікробіологічні основи екології навколишнього середовища.
- Лекція 3.Тема: Мікробіологічні основи екології навколишнього середовища.

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
