Химические свойства карбоновых кислот и их производных.
За счет образования прочных межмолекулярных водородных связей температуры кипения карбоновых кислот значительно выше, чем у соответствующих альдегидов и спиртов. В газовой фазе низшие карбоновые кислоты присутствуют в виде димеров, а в жидкой фазе карбоновые кислоты способны образовывать помимо димерных линейно-цепные структуры за счет водородных связей.
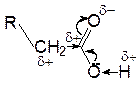 Благодаря влиянию гидроксильного атома кислорода карбонильная активность центрального атома углерода и, следовательно, склонность к реакциям нуклеофильного присоединения сильно понижается. Наоборот, акцепторное влияние карбонильной группы в целом увеличивает поляризацию связи О-Н в гидроксильной группе, что приводит к облегчению диссоциации этой связи по кислотному типу по сравнению со спиртами. рКа карбоновых кислот близко к 5 и степень диссоциации в 0,1 молярном растворе около 1%.
Благодаря влиянию гидроксильного атома кислорода карбонильная активность центрального атома углерода и, следовательно, склонность к реакциям нуклеофильного присоединения сильно понижается. Наоборот, акцепторное влияние карбонильной группы в целом увеличивает поляризацию связи О-Н в гидроксильной группе, что приводит к облегчению диссоциации этой связи по кислотному типу по сравнению со спиртами. рКа карбоновых кислот близко к 5 и степень диссоциации в 0,1 молярном растворе около 1%.
Степень диссоциации (по кислотному типу) карбоновых кислот напрямую зависит от электронных эффектов заместителя при карбонильном атоме углерода и увеличивается при усилении акцепторных свойств данного заместителя (увеличивается частичный положительный заряд на карбонильном атоме углерода, что приводит к дополнительной поляризации связи О-Н). В ароматических карбоновых кислотах степень диссоциации (сила кислоты) в общем случае выше, чем у их алифатических аналогов и зависит от электронных эффектов заместителей в бензольном кольце.
рКа некоторых алифатических и ароматических монокарбоновых кислот в воде при стандартных условиях.
| Карбоновая кислота | рКа | Карбоновая кислота | рКа |
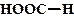
| 3,75 | 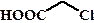
| 2,86 |
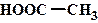
| 4,76 | 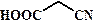
| 2,47 |
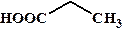
| 4,87 | 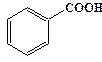
| 4,20 |
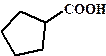
| 4,99 | 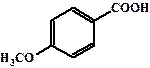
| 4,50 |
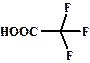
| 0,23 | 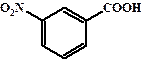
| 3,46 |
Более подробную информацию о значениях рКа карбоновых кислот можно найти в справочнике [«Свойства органических соединений»…] и в книге [Реутов О.А., Курц А.Л., Бутин К.П. «Органическая химия» глава 18].
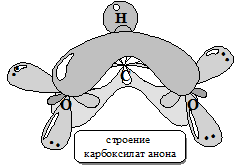 В результате диссоциации, а также в реакциях с основаниями из карбоксильной группы карбоновых кислот образуется симметричный карбоксилат -анион.
В результате диссоциации, а также в реакциях с основаниями из карбоксильной группы карбоновых кислот образуется симметричный карбоксилат -анион.
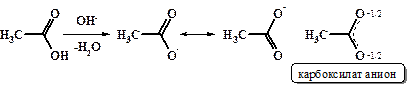 Данные физических исследований подтверждают предположение о симметричности карбоксилат-аниона. В действительности обе связи С-О в карбоксилат-анионе имеют одинаковую длину (1,27 Ǻ для формиат-аниона), что является промежуточным значением между 1,20 Ǻ С=О и 1,34 Ǻ С-О в свободной муравьиной кислоте.
Данные физических исследований подтверждают предположение о симметричности карбоксилат-аниона. В действительности обе связи С-О в карбоксилат-анионе имеют одинаковую длину (1,27 Ǻ для формиат-аниона), что является промежуточным значением между 1,20 Ǻ С=О и 1,34 Ǻ С-О в свободной муравьиной кислоте.
В сильнокислой среде карбоновые кислоты и некоторые их производные проявляют свойства слабых оснований и протонируются по карбонильный группе подобно альдегидам и кетонам.
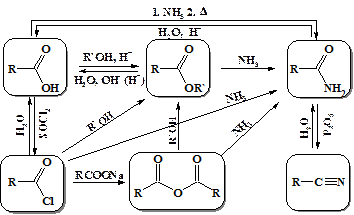 Реакции карбоновых кислот и их производных весьма многообразны. На схеме представлены основные пути взаимных превращений производных карбоновых кислот. Все реакции протекают по типу нуклеофильного присоединения (замещения) при карбонильном атоме углерода.
Реакции карбоновых кислот и их производных весьма многообразны. На схеме представлены основные пути взаимных превращений производных карбоновых кислот. Все реакции протекают по типу нуклеофильного присоединения (замещения) при карбонильном атоме углерода.
Реакционная способность производных карбоновых кислот в реакциях нуклеофильного замещения напрямую зависит от карбонильной активности. Последняя является мерой частичного положительного заряда наведенного на атом углерода (как в альдегидах и кетонах). Рассмотрение электронных эффектов заместителей показывает, что наиболее активными среди производных карбоновых кислот являются галогенангидриды, из них можно получить любые другие производные карбоновых кислот (кроме нитрилов) напрямую, а наименее активными амиды карбоновых кислот, для превращения которых необходим катализ, высокая температура и продолжительное время.
На примере реакции образования сложных эфиров из карбоновых кислот –реакции этерификации (от английского слова ester – сложный эфир) рассмотрим механизм нуклеофильного замещения при карбонильном атоме. В связи с низкой активностью карбоксильной группы, для осуществления реакции этерификации необходим катализ. При катализе сильными кислотами на первой стадии процесса протонируется карбонильный атом кислорода. За счет этого резко облегчается атака нуклеофильными агентами по несущему положительный заряд карбонильному атому углерода. В качестве атакующего нуклеофила в данном случае выступает спирт. Образующийся в процессе атаки четырехзамещенный sp3-гибридизованный интермедиат подвергается обратимому депротонированию и протонированию. В результате отщепления молекулы воды образуется продукт нуклеофильного замещения – сложный эфир.
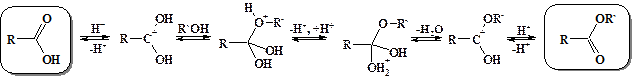
Учитывая равновесность всех стадий данного процесса, он может быть реализован и в обратном направлении. Поэтому вода в присутствии сильных кислот гидролизует сложные эфиры до карбоновых кислот и спиртов. Однако ни образование сложных эфиров, ни их гидролиз в кислой среде не проходит до конца. В условиях щелочного гидролиза сложных эфиров последняя стадия процесса необратима – образуется не проявляющий карбонильной активности карбоксилат анион, и щелочной гидролиз сложных эфиров протекает нацело.
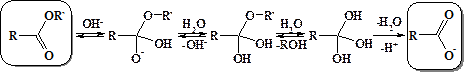
В промышленности реакцию щелочного гидролиза используют в частности для омыления жиров, которые представляют собой сложные эфиры жирных кислот и глицерина. В результате образуется собственно глицерин и мыло – соли жирных кислот. Натриевые соли твердые (т.н. твердые мыла), а калиевые – жидкие (жидкие мыла).
Существует еще несколько механизмов образования и гидролиза сложных эфиров карбоновых кислот, но они весьма редко реализуются на практике. Более подробно о механизмах этерификации и гидролиза эфиров можно посмотреть в [Марч Дж. Органическая химия т.2, стр. 109].
Как видно из вышеприведенной схемы из производных карбоновых кислот обладающих высокой карбонильной активностью – хлорангидридов и ангидридов кислот можно получить все остальные производные без дополнительных условий.
«Жизнь есть способ существования белковых тел»
Ф. Энгельс.
Среди производных карбоновых кислот в природе встречаются некоторые кислоты в свободном виде – муравьиная (муравьи), уксусная (крапива), щавелевая (щавель и т.п.) бензойная (клюква) и другие кислоты. Так же присутствуют сложные эфиры в составе жиров и масел, а также восков и камедей. Особое место в живой материи занимают амиды карбоновых кислот. Помимо свободных амидов, встречающихся в живой материи, за счет образования амидной (пептидной) связи между амино-группой одной α-аминокислоты и карбоксильной группой другой образуется пептидная цепочка из аминокислотных фрагментов (т.н. аминокислотных остатков). Эта цепочка, определенным образом свернутая в пространстве, называется белком. Природные белки имеют молекулярную массу от единиц до нескольких сотен кДа, что соответствует сотням и даже тысячам аминокислотных остатков. Амидная связь оказывается самой прочной из связей О=С–Х. Она не разрушается в слабокислой и умеренно щелочной среде. Полный гидролиз пептидной цепочки можно осуществить, действуя горячей 6Н соляной кислотой в течение нескольких часов. Этот способ разрушения белка используют при определении аминокислотного состава неизвестной пептидной последовательности. Полученную после гидролиза смесь аминокислот, из которых состоял пептид, анализируют с помощью жидкостной хроматографии и определяют качественный и количественный состав исходного белка. Природные белки содержат в своем составе только один из оптических изомеров каждой α-аминокислоты. Как правило, в природных пептидах содержатся L-аминокислоты (см. главу 4), редко встречаются D-аминокислоты.
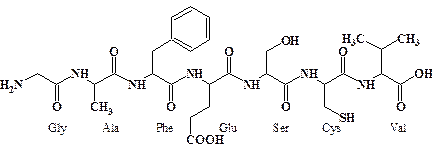 На схеме в качестве примера представлен гептапептид состоящий из последлвательности остатков глицина (Gly), аланина (Ala), фенилаланина (Phe), глютаминовой кислоты (Glu), серина (Ser), цистеина (Cys) и валина (Val). (Конфигурация оптических центров для простоты не показана).
На схеме в качестве примера представлен гептапептид состоящий из последлвательности остатков глицина (Gly), аланина (Ala), фенилаланина (Phe), глютаминовой кислоты (Glu), серина (Ser), цистеина (Cys) и валина (Val). (Конфигурация оптических центров для простоты не показана).
Для большинства природных и некоторых синтетических α-аминокислот используют тривиальные названия и трехбуквенные сокращения (см. главу 16 приложение 1).
Реакции восстановления производных карбоновых кислот.
Аналогично альдегидам и кетонам, карбоновые кислоты и их производные восстанавливаются под действием металлокомплексных гидридов и водорода на катализаторах. Восстановление карбоновых кислот и их сложных эфиров приводит к образованию спиртов, восстановление амидов и нитрилов теми же реагентами позволяет получать амины.
Реакция с реактивами Гриньяра.
 Свободные карбоновые кислоты редко вводят в реакцию с реактивами Гриньяра по причине сильных основных свойств последних. При взаимодействии со свободной кислотой один эквивалент магний-органического соединения расходуется на образование соли кислоты. Для уменьшения количества эквивалентов реактива Гриньяра иногда в реакцию сразу вводят соли карбоновых кислот.
Свободные карбоновые кислоты редко вводят в реакцию с реактивами Гриньяра по причине сильных основных свойств последних. При взаимодействии со свободной кислотой один эквивалент магний-органического соединения расходуется на образование соли кислоты. Для уменьшения количества эквивалентов реактива Гриньяра иногда в реакцию сразу вводят соли карбоновых кислот.
Как правило, для получения кетонов используют хлорангидриды или амиды кислот.
Реакция реактивов Гриньяра со сложными эфирами в большинстве случаев протекает неоднозначно, и основным продуктом вместо ожидаемого кетона часто становится третичный спирт – продукт двойного присоединения магний-органического соединения по карбоксильной группе сложного эфира.
Реакция по a-углеродному атому
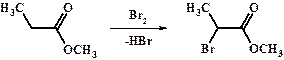 Все карбонильные соединения, за счет акцепторного влияния карбонильной или карбоксильной группы на соседнюю -СН2- и способности образовывать енольную форму с образованием кратной связи С=С, реагируют с галогенами по типу реакции присоединения-отщепления с образованием a-галогенкетонов или a-галогенкислот и a-галогенсложных эфиров.
Все карбонильные соединения, за счет акцепторного влияния карбонильной или карбоксильной группы на соседнюю -СН2- и способности образовывать енольную форму с образованием кратной связи С=С, реагируют с галогенами по типу реакции присоединения-отщепления с образованием a-галогенкетонов или a-галогенкислот и a-галогенсложных эфиров.
За счет высокой нуклеофугной подвижности атома галогена в a-галогенкетонах и сложных эфирах многие a-галогенпроизводные карбонильных соединений проявляют свойства сильных лакриматоров (веществ раздражающих слизистые оболочки и слезоточивых).
Сложноэфирная конденсация.
Аналогично альдегидам и кетонам, сложные эфиры карбоновых кислот вступают в реакции конденсации с участием соседней -СН2- группы с образованием β-кетоэфиров карбоновых кислот. Механизм сложноэфирной конденсации близок к таковому для альдольно-кротоновой. Однако, для успешного проведения сложноэфирной конденсации, необходимо использовать более сильные основания, так как сложные эфиры обладают меньшей карбонильной активностью и гораздо труднее енолизуются.

Реакции декарбоксилирования.
При высокой температуре большинство карбоновых кислот подвергается декарбоксилированию с выбросом СО2. Ароматические карбоновые кислоты отщепляют углекислый газ гораздо легче и при более низких температурах. Особенно легко отщепляют карбоксильную группу арилдикарбоновые и поликарбоновые кислоты. Также легко (ниже 100˚С) декарбоксилируется малоновая кислота. В некоторых случаях, для понижения температуры декарбоксильрования и более полного протекания процесса используют катилизаторы (хромит меди) и соответствующие высококипящие растворители. В качестве примера можно привести получение никотиновой кислоты (пиридин-3-карбоновой) окислением хинолина в пиридин-2,3-дикарбоновую кислоту с последующим селективным декарбоксилированием последней.
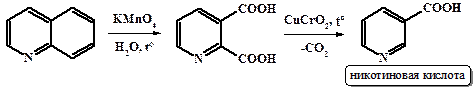
Реакции дикарбоновых кислот
Щавелевая, малоновая и отчасти янтарная кислоты обладают сильно отличающимися от обычных карбоновых кислот свойствами. Более протяженные дикарбоновые кислоты близки по свойствам к соответствующим монокарбоновым с той только разницей, что в молекуле присутствуют два реакционных центра. Последние способны к внутримолекулярным реакциям между концевыми карбоксильными группами. В частности, янтарная (С4), глутаровая (С5) и адипиновая (С6) кислоты способны образовывать циклические ангидриды и амиды, а адипиновая и пимелиновая (С7) кислоты легко вступают в реакцию образования циклических кетонов по реакции разложения соответствующих кальциевых, бариевых, железных или ториевых солей соответствующих кислот (см. получение кетонов). Этот способ широко используется для синтеза циклических кетонов с размером цикла от 4 до 7.
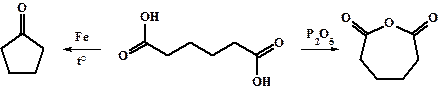
Нагревание янтарной, а особенно малоновой кислот приводит к их декарбоксилированию с образованием пропионовой и уксусной кислот соответственно.
Сложные эфиры глутаровой, адипиновой и пимелиновой кислот способны вступать во внутримолекулярную сложноэфирную конденсацию. Последующий гидролиз и декарбоксилирование приводит к циклическим кетонам.
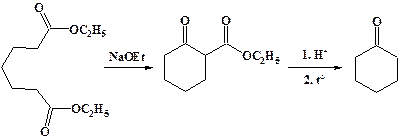
Дата добавления: 2016-06-15; просмотров: 3746;
Поиск по сайту
Узнать еще
- V. СПОСОБЫ ПОЛУЧЕНИЯ И ХИМИЧЕСКИЕ СВОЙСТВА АЦЕТИЛЕНОВЫХ И ДИЕНОВЫХ УГЛЕВОДОРОДОВ
- А) кислотные свойства
- А) Основные свойства
- А) Физические свойства минералов
- А. ОДНООСНОВНЫЕ ПРЕДЕЛЬНЫЕ КИСЛОТЫ
- А. Оптические свойства минералов
- А. Свойства и виды рецепторов. Взаимодействие рецепторов с ферментами и ионными каналами
- Аденилат (адениловая кислота), Гуанилат (гуаниловая кислота),

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
