Електролітична дисоціація. Сильні і слабкі електроліти
Речовини, які проводять електричний струм у водних розчинах, називаються електролітами. Це перш за все солі, кислоти, основи.
Кількісні дослідження залежності властивостей розведених розчинів від концентрації розчиненої речовини показали, що пониження тиску пари над розчином, підвищення температури кипіння, пониження температури кристалізації, осмотичний тиск розчинів описуються одним і тим же законом розведених розчинів Рауля—Вант-Гоффа (1886р.):
Властивості розведених розчинів змінюються прямо пропорційно відносно числа розчинених частинок, тобто молекулярній концентрації розчиненої речовини.
Проте ці закономірності виявились неприйнятними до водних розчинів солей, кислот, основ. У даному випадку зазначені вище показники були завжди вищими, ніж розраховані теоретично на основі закону Рауля—Вант-Гоффа. Це наводило на думку про те, що при розчиненні у воді солей, кислот, основ їх молекули розпадаються на більш дрібні частинки і в результаті з’являється більше частинок, ніж було розчинено молекул. Підвищення температури кипіння, пониження температури плавлення і осмотичний тиск розчинів залежать тільки від числа частинок в розчині і не залежать від їх природи.
На основі вивчення результатів електропровідності розчинів було виявлено, що саме солі, кислоти, основи у водних розчинах добре проводять електричний струм, тобто саме ті речовини, для яких спостерігаються відхилення від закону розведених розчинів.
У 1887 році С. Арреніус запропонував гіпотезу йонізації, яка зв’язала ці особливості солей, кислот, основ з їх електропровідністю в розчинах.
Суть гіпотези йонізації була у тому, що молекули солей, кислот, основ у водному розчині частково розпадаються на самостійні йони. Чим більше таких йонів, тим більша електропровідність розчину. У міру розпаду молекул на йони зростає і загальне число розчинених частинок, так як при цьому з одної частинки утворюється дві чи більше. Отже, закон розведених розчинів справедливий і для водних розчинів солей, кислот, основ, якщо враховувати як самостійні частинки не тільки молекули, але йони, які утворюються при розпаді молекул.
С. Арреніус був прихильником „фізичної” теорії розчинів і не враховував взаємодію розчиненої речовини з розчинником. Він вважав, що молекули розпадаються на вільні йони, які хаотично рухаються у розчині як і молекули газів у газових сумішах.
І.О. Каблуков був переконаний, що у водних розчинах містяться не вільні, а гідратовані йони, причому саме гідратація є основною причиною йонізації молекул.
Гіпотеза йонізації після її експериментального підтвердження отримала назву теорії електролітичної дисоціації.
Уявлення про хаотичне розподілення йонів у розчині не відповідає дійсності, так як не враховує електростатичну взаємодію між йонами. Електростатична взаємодія проявляється на відносно великих відстанях і в розчинах сильних електролітів, які націло розпалися на йони, концентрація йонів значна, відстані між ними невеликі. Тому електростатична взаємодія між йонами впливає на їх розподілення у розчині. Виникає впорядковане розподілення аналогічне розподіленню йонів у йонних кристалах, де кожен йон оточений йонами протилежного знаку заряду.
Таким чином, розподілення йонів буде визначатися співвідношенням електростатичної енергії і енергії хаотичного руху йонів.
Електростатичні сили прагнуть створити таке розподілення, при якому кожний йон оточений тільки йонами протилежного знаку заряду, але цьому протидіє хаотичний рух йонів, що призводить до безладного розподілення. Наслідком цього є те, що навколо кожного йона утворюється своєрідна йонна атмосфера, в якій переважають йони протилежного у порівнянні з центральним йоном знаку заряду.
У 1923р. голландський фізик П. Дебай і німецький хімік-теоретик Е. Гюккель розробили теорію сильних електролітів. За цією теорією йони розподілені в об’ємі розчину (в кожний даний момент) не хаотично, а у відповідності із законом їх кулонівської взаємодії. З цього положення методом статистичної фізики знайдено розподілення йонів різних знаків заряду навколо кожного окремого йона. Таким чином, відкрито існування йонної атмосфери, яка є навколо кожного йона і яка складається з йонів протилежного центральному йону знаку заряду.
Це статистично нерівномірне розподілення у просторі електричних зарядів різних знаків зв’язано з потенціальною енергією їх взаємного тяжіння, яка входить як складова частина у величину ізобарного потенціалу розчину. Розрахувавши енергію взаємного притяжіння йонів, можна краще зрозуміти всі термодинамічні властивості електролітів і визначити ряд властивостей розведених розчинів електролітів.
Виходячи з теорії електролітичної дисоціації, солі можна визначити як сполуки, що у водних розчинах дають йони металу і кислотного залишку; кислоти – сполуки, що дають у водних розчинах гідроген-іони (точніше йони гідроксонію Н3О+); основи – сполуки, що дають у водних розчинах гідроксид-іони. Сполуки, молекули яких при одних і тих же умовах здатні відщеплювати і гідроген-іони, і гідроксид-іони, називаються амфотерними.
Слід зазначити, що крім води йони утворюються при розчиненні речовин в спиртах, оцтовій кислоті, рідкому амоніаку.
Електролітична дисоціація є процес зворотний. У кожний момент за рахунок дисоціації молекул утворюються йони, а в результаті зіткнення йонів – молекули:
дисоціація D моляризація.
У результаті встановлюється рівновага між недисоційованими молекулами електролітів і йонами, що утворилися при їх дисоціації.
Зміна концентрації розчину відбивається на процесах дисоціації і моляризації. Розведення розчину суттєво не збільшує швидкість дисоціації, а швидкість моляризації при цьому зменшується внаслідок того, що вона залежить від числа зіткнень між різнойменними йонами, яких при розведенні розчину в одиниці об’єму стає менше. В результаті рівновага зміщується і ступінь дисоціації електроліту при розведенні розчину зростає.
Теорія дисоціації прийнятна лише для слабких електролітів, які являють собою нейонізовані сполуки, що утворюють йони лише при розчиненні. Сильні електроліти, серед яких більшість солей, складаються із йонів у всіх агрегатних станах. При їх розчиненні чи розплавленні відбувається тільки розділення йонів – йонізація, а не дисоціація, оскільки вони не складаються з молекул. Кристал солі, наприклад NaCl, можна розглядати як одну молекулу, у вузлах кристалічної ґратки якого розміщені йони Na+ і Cl–. Формула NaCl відображає тільки найпростіше співвідношення між йонами в натрій хлориді.
Йони протилежних знаків заряду, які складають йонну кристалічну ґратку, зв’язані між собою великими електростатичними силами. При переході йонів у розчин енергія електростатичної взаємодії між йонами в кристалічній ґратці протиставляється енергії взаємодії йонів з дипольними молекулами розчинника, який витягує йони з ґратки в розчин. При цьому йони оточуються молекулами розчинника, які утворюють навколо йона сольватну (якщо розчинник вода – гідратну) оболонку.
Якщо енергія взаємодії йонів з розчинником стає порівняльною з енергією йонів, що коливаються біля стану рівноваги у кристалічній ґратці, то відбувається розчинення з йонізацією.
Взаємодія дипольних молекул розчинника з елементами кристалічної ґратки може призвести до утворення електроліту навіть при розчиненні речовин, що мають молекулярну кристалічну ґратку або ґратку проміжного типу. При цьому відбувається розчинення з дисоціацією.
До розчинів сильних електролітів не можна застосовувати правило константи рівноваги. Отже, йонізація сильних електролітів є не рівноважний процес і її, на відміну від дисоціації слабких електролітів, треба виражати з допомогою рівнянь незворотної реакції:
NaOH = Na+ + OH–;
H2SO4 = 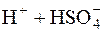 ;
;
ZnCl2 = Zn2+ + 2Cl–.
Багатоосновні кислоти, як правило, дисоціюють ступінчасто. Перший гідроген-іон відривається завжди легше, ніж наступні. Тому константи дисоціації другої і третьої стадії дисоціації значно менші, ніж першої. Наприклад, сульфатна кислота у першій стадії дисоціації виступає як сильний електроліт, а в другій – як електроліт середньої сили:
HSO4– D H+ +SO42–
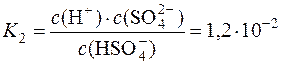 .
.
Саме через це сульфатна кислота порівняно легко утворює кислі солі.
Фосфатна кислота є електролітом середньої сили. У водному розчині навіть перша константа дисоціації цієї кислоти є мала:
H3PO4 DH+ + H2PO4–;
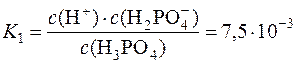 ,
,
а наступні константи дисоціації ще менші: K2=6,2·10–8; K3=2,2·10–13. Отже, у водному розчині фосфатної кислоти йонів 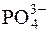 міститься надзвичайно мало, практично за третьою стадією фосфатна кислота майже не дисоціює. Зміщення рівноваги реакції дисоціації
міститься надзвичайно мало, практично за третьою стадією фосфатна кислота майже не дисоціює. Зміщення рівноваги реакції дисоціації
HPO42– D H+ + PO43–
у бік утворення йонів відбувається лише за умови зв’язування йона Н+ у лужному середовищі.
До слабких електролітів відносяться оцтова кислота (K=1,75·10–5), ціанідна (K=7,2·10–10), карбонатна (K1=4·10–7), боратна (K1=5,3·10–10), сульфідна (K1=8,7·10–8) та ін.
З розчинних у воді основ сильними є LiOH, NaOH, KOH, RbOH, CsOH, Ba(OH)2, Sr(OH)2, які виступають як сильні електроліти. Амоній гідроксид є слабким електролітом (K=7,2·10–7). Слабким електролітом є також AgOH (K=1,1·10–4).
У вільному стані кислоти не йонізовані і не проводять електричний струм. Так, рідкий HBr погано проводить електричний струм, але його водний розчин проводить електричний струм добре. Розчин сухого газоподібного HCl у бензені не проводить електричний струм і не реагує з металічним цинком. Отже, реакція цинку з водним розчином гідроген хлориду у дійсності є реакція цинку з йонами H+, що є в розчині.
Обмеження теорії електролітичної дисоціації виявляється у тому, що вона розглядає як кислоти і основи лише ті речовини, які утворюють йони H+ чи ОН– у водних розчинах. Теорія не уточнює, чи є, наприклад, сухий HCl кислотою, чи він стає кислотою тільки при розчиненні у воді. З іншого боку, NH3 може утворювати ОН––іон тільки в результаті взаємодії з водою:
NH3 + H2O D NH4OH D NH4+ + ОН–.
Однак немає жодного експериментального доказу існування нейонізованої речовини з формулою NH4OH – амоній гідроксид. Так що ж являє собою розчин амоніаку у воді? Точніше було б писати NH3·H2O.
Відомі також сильні основи, в розчинах яких немає гідроксильних йонів (С2Н5О–+Na+; 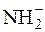 +Na+).
+Na+).
У концентрованих розчинах сильних електролітів спостерігаються дещо менші пониження температури кристалізації або збільшення осмотичного тиску, ніж це повинно бути при повній їх йонізації. Проте з цього не слід робити висновки, що при збільшенні концентрації розчинів сильні електроліти стають слабшими. Кількість йонів сильних електролітів у концентрованих розчинах не стає меншою. Всі властивості не дуже розведеного розчину електроліту, що залежать від концентрації йонів, вказують на зменшення ефективної концентрації йонів. Тому для характеристики властивостей йонів слід користуватися їх активністю, а не концентрацією. Активність йона пропорційна його ефективній концентрації:
а=f·c.
Оскільки активність йона менша за його концентрацію, то коефіцієнт активності f менший одиниці. Тільки у дуже розведених розчинах він наближається до одиниці.
У розведених розчинах сильних електролітів різні їх властивості адитивні, тобто величини властивостей електроліту є сумою відповідних властивостей йонів. У концентрованих же розчинах правило адитивності не витримується. Для концентрованих розчинів сильних електролітів теорія сильних електролітів Дебая-Гюккеля виявляється малоефективною.
Слабкі електроліти дисоційовані на йони лише частково і йонна сила таких розчинів незначна навіть при великих концентраціях розчинів. Отже, при розрахунках рівноваги реакцій дисоціації слабких електролітів без великої помилки можна користуватися значеннями концентрацій йонів і молекул замість їх активностей. Якщо ж розчин являє собою суміш електролітів, то йонна сила, яка залежить від загальної концентрації йонів, може бути настільки великою, що для розрахунку рівноваги необхідно переходити від аналітичних концентрацій до активностей навіть для тих йонів, концентрація яких незначна. У вираз константи рівноваги достатньо ввести лише активності йонів, оскільки активність недисоційованих молекул в дуже незначній мірі залежить від йонної сили.
Концентрацію йонів (моль/дм3), знаходять як добуток концентрації електроліту (моль/дм3), ступінь дисоціації α і коефіцієнта z, який показує, скільки йонів даного типу утворюється при розпаді одної молекули.
При дисоціації слабкого електроліту установлюється рівновага між недисоційованими молекулами і йонами.
Розглянемо, як приклад, дисоціацію оцтової кислоти:
СН3СООН D Н+ + СН3СОО–.
Виходячи із закону діючих мас, константа дисоціації матиме вираження:
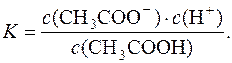
Константа дисоціації K – це відношення добутку молярних концентрацій йонів слабкого електроліту до молярної концентрації слабкого електроліту у стані рівноваги за даної температури. Чим менша K, тим буде меншою концентрація йонів і більшою концентрація недисоційованих молекул, тобто тим слабкішим є електроліт.
Ступінь дисоціації α є відношення числа молекул, що розпалися на йони, до загального числа всіх взятих молекул слабкого електроліту.
Між ступенем дисоціації і константою дисоціації є залежність.
Якщо с – молярна концентрація слабкого електроліту, тоді у стані рівноваги концентрації продуктів дисоціації дорівнюють с·α, а недисоційованих молекул – (с–с·α). Підставляючи ці значення у вираження константи дисоціації, маємо:
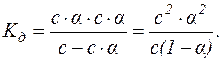
Для слабких електролітів α – дуже мала величина, тоді (1–α) можна вважати за 1.
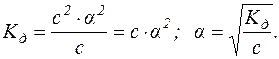
Цей вираз є законом розведення В. Оствальда, у якому дається залежність між α, Kд і загальною концентрацією слабкого електроліту. Із цього закону випливає, що ступінь дисоціації слабкого електроліту збільшується при розведенні його розчину.
Часто буває необхідним зробити розчин слабкої кислоти чи слабкої основи ще менш кислим, чи лужним, не застосовуючи реакцію нейтралізації.
Виходячи із закону діючих мас до процесу електролітичної дисоціації, щоб зменшити концентрацію Н+ (наприклад, при дисоціації СН3СООН), треба збільшити концентрацію СН3СОО– введенням у розчин добре дисоційованої солі оцтової кислоти (наприклад NaСН3COO). Понизити дисоціацію, наприклад NH4ОН, можна додаванням до розчину сильної основи чи добре дисоційованої солі амонію (наприклад NH4Сl).
Отже, шляхом додаткового введення у розчин одного із однойменних йонів можна пригнічувати дисоціацію цього електроліту і, таким чином, створити необхідні оптимальні умови проведення реакції.
Дата добавления: 2020-10-14; просмотров: 775;
Поиск по сайту
Узнать еще

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
