Патогенез опухолевого роста (онкогенез)
Современные концепции онкогенеза связаны с успехами молекулярной биологии и генной инженерии. В процессе опухолевого превращения клетка проходит несколько стадий, приобретая на каждой из них новые свойства. В основе онкогенеза лежат изменения особых генов: протоонкогенов и/или антионкогенов.
В нормальном состоянии эти гены играют ключевую роль в важнейших проявлениях жизнедеятельности клетки, осуществляя позитивный (протоонкогены) или негативный (антионкогены) контроль клеточного деления, регуляцию организации цитоскелета, а также участвуя в механизмах программируемого «самоубийства» клетки - апоптоза. Однако различные мутации могут привести к усилению или извращению функций протоонкогенов (с превращением их в онкогены) и/или к инактивации функции антионкогенов. Такие изменения в геноме приводят клетку к опухолевой трансформации.
Протоонкогены- специфические гены нормальных клеток, которые осуществляют позитивный контроль процессов пролиферации и мембранного транспорта.Под влиянием мутаций протоонкогены претерпевают так называемую активацию, что способствует их превращению в онкогены, экспрессия которых вызывает возникновение и прогрессию опухолей. Протоонкоген может превратиться в онкоген при замене в нем даже одного из 5000 нуклеотидов. В настоящее время известно около сотни протоонкогенов. Нарушение функций протоонкогенов вызывает их превращение в онкогены и способствует опухолевой трансформации клетки. Онкогены обозначают соответственно их нахождению: в вирусе (v - virus) или в клетке (c - cellula), а также по характеру опухоли (src, sis, myc, erb и т.д.); например, v-src - вирус саркомы Рауса; c-myc - клеточный онкоген миелоцитарного лейкоза; c-erb - клеточный онкоген эритробластоза; v-sis - вирус саркомы обезьян.
Продуктами протоонкогенов являются белки, которые имеют различную локализацию в клетке. Белки, кодируемые протоонкогенами src и ras, связаны с клеточной мембраной; erbB или fmc - с плазматической и внутриклеточной мембранами; erbA или mos расположены в цитоплазме; myc, fos, jun или myb - в клеточном ядре; sis секретируется во внешнюю среду. Белки, кодируемые протоонкогенами, непосредственно участвуют в проведении ростстимулирующих сигналов, побуждающих клетку к делению. В литературе достаточно подробно описана цепь последовательных сигналов для нормального роста и функционирования клеток. Все ключевые белки, участвующие в этой цепи, кодируются протоонкогенами. Например, рецептор эпидермального фактора роста кодируется протоонкогеном erbB, тирозиновая протеинкиназа на внутренней поверхности плазматической мембраны клетки - протоонкогеном src, G-белки - протоонкогеном семейства ras,
ядерные транскрипционные факторы являются продуктами протоонкогенов fos, jun, myc.
В результате мутационного превращения протоонкогенов в онкогеныодно или несколько звеньев этой цепи спонтанно становятся сверхактивными. Так, например, аномальная форма рецептора эпидермального фактора роста (ЭФР) приводит к тому, что такой рецептор постоянно находится в состоянии активации, посылая ложные сигналы, стимулирующие размножение клеток. G-белки, утратившие способность гидролизовать гуанидинтрифосфат в гуанидиндифосфат, остаются постоянно связанными с гуанидинтрифосфатом и непрерывно передают ростстимулирующий сигнал. Связанные с плазматической мембраной и цитоплазматические протеинкиназы могут стать постоянно активированными без необходимости в стимуляции. Может резко возрасти количество или повыситься функциональная активность транскрипционных факторов, влияющих на экспрессию генов, побуждающих клетку к делению. Следствием этих нарушений является неконтролируемое деление клеток, что характеризует опухоли. Поэтому считается, что протоонкогены осуществляют позитивную регуляцию процесса пролиферации. Блокирование же любого из этапов передачи митогенного сигнала может в принципе привести к нарушению регуляции пролиферации опухолевых клеток и потенциально к торможению роста опухоли. В эксперименте уже исследовано достаточно большое количество препаратов, влияющих на вышеперечисленные процессы. Большинство из них находится на стадии предклинического изучения, хотя ряд препаратов уже прошли первую фазу клинических испытаний и показали себя достаточно эффективными при некоторых видах опухолей.
Выделяют несколько механизмов трансформации протоонкогена в онкоген:1) инсерционная активация - активация протоонкогена клетки при включении в ее геном вирусного промотора; 2) амплификация - в результате умножения (копирования) числа одинаковых протоонкогенов, которые, как и в первом случае, приводят к усилению синтеза онкобелков (так называемый эффект дозы), при этом могут появляться добавочные участки хромосомы; 3) активация протоонкогена при транслокации участка хромосомы в другое место той же или другой хромосомы; 4) точечная мутация протоонкогена, приводящая к синтезу онкобелков, которые нарушают регуляцию клеточного деления.
Описанные нарушения генома могут быть вызваны различными канцерогенными факторами: химическими, физическими (уль-
трафиолетовое и ионизирующее излучение) и др. В дальнейшем при прогрессировании опухоли частота мутаций, транслокаций и других хромосомных нарушений, выявляемых в опухолевых клетках, значительно возрастает, и это является следствием утраты контроля над стабильностью генома, что свойственно опухолевым клеткам.
Мутация обычно возникает в одной из двух копий (аллелей) протоонкогена, располагающихся в парных хромосомах, и проявляется как доминантная. Таким образом, одной мутации достаточно для превращения протоонкогена в действующий онкоген. Для многих типов опухолей характерна гиперэкспрессия онкогенов семейства ras в результате точечных мутаций. Так, мутация гена k-ras отмечается в 60-80% случаев рака поджелудочной железы, и поэтому обнаружение этой мутации имеет диагностическое значение. В клетках плоскоклеточной карциномы обнаруживается гиперэкспрессия онкогена erbB-1 в результате амплификации. Для хронического миелоидного лейкоза характерна транслокация гена abl с 9-й на 22-ю хромосому с укорочением последней (так называемая филадельфийская хромосома). Лимфома Беркитта в значительной мере обусловлена перемещением протоонкогена myc на другую хромосому и попаданием под его контроль энхансера гена, кодирующего цепи иммуноглобулина. В результате клетка вместо «включения» генов для синтеза антител «включает» онкоген myc, что усиливает пролиферацию. Вызванное транслокацией (между 15-й и 17-й хромосомами) соединение части протоонкогена pml с геном, кодирующим клеточный рецептор ретиноевой кислоты, ведет к образованию гибридного гена, обусловливающего развитие острого промиелоцитарного лейкоза. Эта транслокация служит диагностическим признаком данного заболевания. В табл. 13-2 представлены данные исследований о некоторых изменениях протоонкогенов, характерных для новообразований человека.
Изменения в геноме клетки могут быть также вызваны онкогенными вирусами. РНК-содержащие вирусы содержат онкогены, образовавшиеся, по-видимому, из клеточных протоонкогенов, захваченных когда-то вирусами. В случае заражения ретровирусами в клетку вносится готовый онкоген. Одним из механизмов опухольтрансформирующего действия ДНК-содержащих вирусов является способность некоторых белков, кодируемых специфическими генами этих вирусов, инактивировать антионкогены в клетках.
Таблица 13-2.Некоторые изменения протоонкогенов, характерные для новообразований у человека (по Б.П. Копнину, 2000)
| Протоонкогены | Функция белка | Изменения | Новообразования |
| RET (GDNF-R) | Рецепторная тирозинкиназа | Точечные активирующие мутации. Рекомбинации, образующие химерные гены Ret/ ptc, кодирующие постоянно активированный рецептор | Синдромы множественных эндокринных неоплазий (MEN2a, MEN2b), медуллярный и папиллярный рак щитовидной железы* |
| ERBB1 (EGF-R) | Рецепторная тирозинкиназа | Амплификация и гиперэкспрессия гена | Глиобластомы и другие нейрогенные опухоли |
| ERBB2 (HER2) | Рецепторная тирозинкиназа | Амплификация и/ или гиперэкспрессия гена | Рак молочной железы |
| PGDF-Rb | Рецепторная тирозинкиназа | Хромосомные транслокации, образующие химерные гены TEL/PDGF-Rb, CVE6/ PDGF- Rb, кодирующие постоянно активированные рецепторы | Хронический миеломоно- цитарный лейкоз, острый миелобластный лейкоз |
| SRC | Нерецепторная тирозинкиназа | Мутации в кодоне 531, отменяющие негативную регуляцию киназной активности | Часть опухолей толстого кишечника на поздних стадиях |
| K-RAS, N-RAS, H-RAS | Участвуют в передаче митогенных сигналов и регуляции морфогенетических реакций | Мутации в кодонах 12, 13, 61, вызывающие образование постоянно активированной GTP-связанной формы ras | 60-80% случаев рака поджелудочной железы; 25-30% различных солидных опухолей и лейкозов |
Продолжение табл. 13-2
| PRADI/ циклин D1 | Регулирует клеточный цикл | Амплификация и/или гиперэкспрессия гена | Рак молочной и слюнных желез |
| C-MYC | Фактор транскрипции, регулирует клеточный цикл и активность теломеразы | Хромосомные транслокации, перемещающие ген под контроль регуляторных элементов генов иммуноглобулинов. Амплификация и/ или гиперэкспрессия гена; мутации, стабилизирующие белок | 1. Лимфома Беркитта 2. Многие формы новообразований |
| CTNNB1 (β-катенин) | Транскрипционный фактор, регулирует с-MYC и циклин D1. Связываясь с кадхерином, участвует в образовании адгезионных контактов | Мутации, увеличивающие количество несвязанного с Е-кадхерином β-катенина, который функционирует как транскрипционный фактор | Наследственный аденоматозный полипоз толстой кишки; различные формы спорадических опухолей |
| BCL2 | Подавляет апоптоз, регулируя проницаемость митохон- дриальных и ядерных мембран | Хромосомные транслокации, перемещающие ген под контроль регуляторных элементов генов иммуноглобулинов | Фолликулярная лимфома |
| ABL | Регулирует клеточный цикл и апоптоз | Хромосомные транслокации, ведущие к образованию химерных генов BCR/ABL, продукты которых стимулируют пролиферацию клеток и подавляют апоптоз | Все хронические миелоидные лейкозы, часть острых лимфобластных лейкозов |
| Окончание табл. 13-2 | |||
| MDM2 | Инактивирует опухолевые супрессоры р53 и pRb | Амплификация и/ или гиперэкспрессия гена | Часть остеосарком и сарком мягких тканей |
* Выделены наследственные формы заболеваний, возникающие при мутациях в половых клетках; в остальных случаях мутации происходят в соматических клетках, которые образуют опухоли.
Антионкогены - гены-супрессоры клеточного деления.Их известно около двух десятков, они действуют как ингибиторы проведения рострегулирующих сигналов в клетке и тем самым предупреждают возможность нерегулируемой пролиферации. Поэтому считается, что антионкогены осуществляют негативную регуляцию пролиферации. Инактивация антионкогенов, вызванная их мутациями (точковыми мутациями и делециями), приводит к неконтролируемому росту клеток. Для выключения антионкогена необходимы две мутации в обоих его аллелях (так как антионкогены - рецессивны), тогда как для превращения протоонкогена в действующий онкоген достаточно только одной (доминантной) мутации. Наличие первой мутации в одном из аллелей антионкогенов предрасполагает к возникновению опухоли, и если такой мутантный аллель унаследован, то достаточно второй мутации, чтобы произошла опухолевая трансформация.
Антионкогены осуществляют роль негативных регуляторов прохождения клетки по клеточному циклу, конечным результатом которого является митоз. Под клеточным циклом понимается упорядоченная последовательность событий от одного клеточного деления до другого. Как известно, клеточный цикл разделяется на 4 дискретных временных периода: G1, S, G2 и М. В фазе S (синтетическая фаза) происходит репликация ДНК; фаза М - митоз; G1 и G2 - промежутки соответственно между М и S и между S и М. Временной механизм прохождения клеткой этого цикла контролируется синтезом и распадом специальных белков - циклинов. Их экспрессия периодически возрастает в течение одной фазы клеточного цикла и затем снижается в другой фазе. Циклины группы В накапливаются в G2-фазе и распадаются в М, циклины Е и D действуют в G1-фазе, циклины А - в S-фазе. Циклины образуют комплексы с так называемыми циклинзависимыми протеинкиназами (ЦЗПК) разных типов. Образование комплексов циклина Е
с ЦЗПК-2 и циклина D с ЦЗПК-4 вызывает фосфорилирование нескольких белков, необходимых для вхождения клетки в S-фазу цикла. Одним из таких белков является продукт антионкогена Rb, с интактивацией которого связано развитие ретинобластомы в раннем детском возрасте и наследственной формы остеосаркомы. Когда этот белок не фосфорилирован, он связан с транскрипционными факторами, которые включают гены, регулирующие репликацию ДНК в S-фазе. Связь этих транскрипционных факторов с белком антионкогена Rb лишает их активности, что препятствует дальнейшему продвижению по циклу клеток, находящихся в G1-фазе. При фосфорилировании белка Rb транскрипционные факторы освобождаются и вызывают вступление клетки в S-фазу. Таким образом, в норме антионкоген Rb осуществляет негативный контроль пролиферации, разрешая или не разрешая клеткам вхождение в фазу репликации ДНК (S-фазу клеточного цикла). Утрата этой функции в результате инактивации антионкогена Rb (его мутации) приводит к тому, что транскрипционные факторы остаются несвязанными, и клетка безостановочно «пробегает» по циклу даже в тех случаях, когда действует запрет на пролиферацию.
Другим антионкогеном, выполняющим в клетке рострегулирующую функцию, является p53. Белок, кодируемый этим антионкогеном, локализован в ядре и является транскрипционным фактором, который включает ряд генов, в том числе и ген waf1. Продукт последнего инактивирует комплексы циклина Е с ЦЗПК-2 и циклина D - с ЦЗПК-4, необходимые для вхождения клетки в S-фазу цикла, в результате чего клетка задерживается в G1-фазе. Мутация антионкогена р53, как и Rb, приводит к нарушению этой регуляции и к безостановочному делению клетки. В норме антионкоген р53 препятствует вхождению в S-фазу клеток с поврежденной или измененной ДНК, поддерживая таким образом целостность клеточного генома. Эту функцию он реализует двумя способами: 1) временно задерживая клетку в G1-фазе цикла, давая ей возможность исправить повреждения ДНК прежде, чем клетка вступит в S-фазу; 2) запуская механизм апоптоза (программируемой гибели клетки) в тех случаях, когда повреждения ДНК столь серьезны, что не подлежат исправлению. Индукция апоптоза в аномальных клетках - чрезвычайно важная функция антионкогена р53. Благодаря апоптозу исключается возможность передачи серьезных поломок ДНК в поколениях клеток. В случае нарушения этой функции р53
происходит накопление клеток с различными хромосомными повреждениями, что является характерным для клеток опухолей.
Мутации антионкогена р53 выявляются примерно в 60% злокачественных опухолей у человека. У людей с врожденной мутацией одного из аллелей р53 опухоли (саркомы, лимфолейкозы, рак молочной железы) обнаруживаются в молодом возрасте с вероятностью 100% (семейный синдром Ли-Фраумени). Антионкоген р53 во многом определяет реакции опухоли на химио- и/или лучевую терапию. В тех опухолях, где р53 не изменен и нормально функционирует, повреждение ДНК под влиянием химиотерапии или облучения вызывает апоптоз опухолевых клеток. В опухолях с инактивированным р53 индуцированные повреждения ДНК не приводят к апоптозу, и такие опухоли резистентны к химио- и лучевой терапии.
Из других антионкогенов, с инактивацией которых связывают возникновение определенных опухолей, можно назвать АРС (семейный аденоматозный полипоз толстого кишечника), WT1 (опухоль Вильмса), NF1 (нейрофиброматоз Реклингаузена) и некоторые другие, которые представлены в табл. 13-3.
Таблица 13-3.Формы опухолей человека, возникающие при инактивации некоторых опухолевых супрессоров и мутаторных генов (по Б.П. Копни-
ну, 2000)
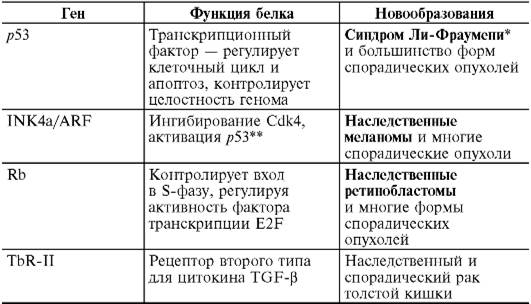 Продолжение табл. 13-3
Продолжение табл. 13-3
| SMAD2, SMAD3 | Передают сигнал от активированных рецепторов TGF-β к Smad 4 | Рак толстой кишки, легкого, поджелудочной железы |
| SMAD4/DPC4 | Транскрипционный фактор - опосредует действие цитокина TGF-β, приводящее к активации ингибиторов Cdk-p21wafl, p27kipl, pl5ink4b | Ювенильный гамартоматозный полипоз желудка и кишечника и различные формы спорадических опухолей |
| Е-кадхерин | Участвует в межклеточных взаимодействиях, инициирует передачу сигналов, активирующих p53, p27kipl | Наследственный рак желудкаи многие формы спорадических опухолей |
| APC | Связывает и разрушает цитоплазматический β-катенин, препятствует образованию транскрипционных комплексов β-катенин/ Tcf | Наследственный аденоматозный полипози спорадические опухоли толстой кишки |
| VHL | Подавляет экспрессию гена VEGF (фактора роста эндотелия сосудов) и других генов, активируемых при гипоксии | Синдром фон Хиппеля- Линдау (множественные гемангиомы), светлоклеточные карциномы почки |
| WT1 | Транскрипционный фактор - связываясь с p53, модулирует экспрессию p53- респонсивных генов | Наследственные нефробластомы(опухоль Вилмса) |
| PTEN/MMACI | Фосфатаза; стимулирует апоптоз, подавляя активность PI3K-PKB/ Akt сигнального пути | Болезнь Коудена (множественные гамартомы)и многие спорадические опухоли |
| NF1 (нейрофибромин) | Белок семейства GAP - переводит онкоген ras из активной в неактивную форму | Нейрофиброматоз первого типа |
Окончание табл. 13-3
| NF2 (мерлин) | Участвует во взаимодействиях мембраны с цитоскелетом | Нейрофиброматоз второго типа: спорадические менингиомы, мезотелиомы и др. опухоли |
| BRCA1 | Повышает активность p53 и других факторов транскрипции, связываясь с RAD51, участвует в узнавании и/или репарации повреждений ДНК | Наследственные опухоли молочной железы и яичникови различные формы спорадических опухолей |
| BRCA2 | Транскрипционный фактор с активностью гистоновой ацетилтрансферазы; связываясь с RAD51, участвует в репарации ДНК | Наследственные опухоли молочной железы и яичникови различные формы спорадических опухолей |
| MSH2, MLH1, PMS1, PMS2 | Репарация неспаренных участков ДНК (mismatch repair) | Неполипозный рак толстой кишки и яичникови многие спорадические опухоли |
* Выделены наследственные формы заболеваний, возникающие при мутациях в половых клетках; ** - локус INK4a/ARF кодирует 2 белка: p16INK4a - ингибитор циклинзависимых киназ Cdk4,6 и p19ARF (Alternative Reading Frame) - продукт альтернативной рамки считывания, который, связывая p53 и Mdm2, блокирует их взаимодействие и препятствует деградации p53. Делеции и многие точечные мутации в локусе INK4a/ARF вызывают одновременно инактивацию супрессоров обоих этих белков.
Таким образом, для появления опухоли необходимо, чтобы в одной и той же клетке возникли мутации в нескольких разных протоонкогенах (с превращением их в онкогены) и антионкогенах. Опухоли одного и того же клеточного или тканевого типа могут развиться в результате различных комбинаций мутаций в протоонкогенах и антионкогенах. Следствием этих генетических изменений является утрата контроля над клеточной пролиферацией. В итоге клетка приобретает трансформированный фенотип, включающий в себя не только нерегулируемую пролиферацию, но и целый ряд характерных изменений структуры и обмена веществ.
Стадийность опухолевого процесса.Предполагается, что опухоль развивается из единичной клетки, которая в процессе роста опухоли проходит ряд стадий.
На I стадии (стадия инициации)под влиянием канцерогенных факторов из одной нормальной исходной клетки в результате стойкого необратимого нарушения генетического материала образуется одна трансформированная клетка. Этот процесс называется опухолевой трансформацией. Канцерогены взаимодействуют с локусами ДНК, содержащими гены, которые регулируют пролиферацию (протоонкогены и антионкогены). Происходит экспрессия протоонкогенов, преобразующихся в онкогены, и инактивация антионкогенов. Вследствие этого клетка становится иммортализованной и потенциально способной к неограниченному делению, но для этого требуется ряд дополнительных условий.
На II стадии (стадия промоции, или активации)происходит активация транформированной клетки под влиянием промотора и последующее превращение ее в активную, пролиферирующую опухолевую клетку. Промоторы - это вещества, которые не являются канцерогенами, не повреждают ДНК, но их воздействие стимулирует пролиферацию уже имеющихся трансформированных клеток. Главное в промоции - стимуляция клеточного деления, вследствие чего создается критическая масса инициированных клеток. К промоторам относятся фенол, скипидар, карболовый эфир и др.
Присутствие промотора, однако, необязательно для индукции опухоли. Он необходим только в случае действия так называемого неполного канцерогена или полного канцерогена, но используемого в таких низких дозах, которые обычно не вызывают опухоли. Полный канцероген, действующий на ткани в достаточно высокой дозе, обладает как инициирующим, так и промоцирующим действием. Неполными канцерогенами считаются те вещества, у которых бластомогенные свойства проявляются только после действия промоцирующего агента. В отличие от инициирующих агентов эффект промоторов может быть обратим, особенно на ранних стадиях формирования опухоли. В течение промоции инициированная клетка приобретает фенотипические свойства трансформированной клетки в результате изменений генной экспрессии (эпигенетический механизм). Однако для индукции опухоли необходимо длительное и непрерывное воздействие промотора. Следовательно, промоторы могут представлять такую же опасность для индукции опухолей, как и полные канцерогены. Так, например, промотор
н-додекан может усиливать канцерогенность бенз(а)пирена в 1000 раз. Фаза промоции в отличие от стадии инициации обратима, по крайней мере на раннем этапе неопластического процесса.
Основные положения канцерогенеза можно обобщить следующим образом:
1) для индукции опухоли недостаточно воздействия одного инициатора (канцерогена) или одного промотора;
2) действия инициатора и промотора не перекрываются во времени;
3) вероятность возникновения опухоли увеличивается в том случае, если промотор действует после инициатора, а не наоборот;
4) интервал между воздействием инициатора и промотора не влияет на возникновение опухоли;
5) индукция опухоли зависит лишь от дозы инициатора.
На III стадии (стадия прогрессии опухоли)идет естественный отбор сильнейших клонов - клональная селекция.
В возникновении опухолевого процесса важное значение имеет возрастная и половая реактивность. С возрастом скорость репарации ДНК снижается и одновременно увеличивается частота мутаций и хромосомных аберраций, особенно при наличии сопутствующих заболеваний и состояний, таких, как хронический стресс, атеросклероз, гипертоническая болезнь, сахарный диабет. Опухоли чаще развиваются в возрасте старше 50 лет, что объясняется увеличением продолжительности действия (суммирование во времени) внешних онкогенных факторов и снижением противоопухолевой резистентности организма. Кроме того, полагают, что в процессе старения возникают гормональные нарушения, способствующие канцерогенезу, причем это связано, по всей видимости, не с недостатком самих гормонов, а со снижением чувствительности центрального (гипоталамо-гипофизарного) звена к действию соответствующего периферического гормона по механизму отрицательной обратной связи. Поэтому не только введение гормонов с лечебной целью, но и нарушение гормонального гомеостаза, вызванное изменениями в функционировании нейроэндокринной системы, способствует развитию рака. Так, повышение концентрации фолликулостимулирующего гормона в крови с возрастом приводит к увеличению частоты возникновения опухолей яичников. Относительное преобладание эстрогенов над прогестероном в период предменопаузы способствует развитию рака молочной железы и эндометрия.
Дата добавления: 2016-07-11; просмотров: 3619;
Поиск по сайту
Узнать еще
- I.6.2 ГИДРОАЭРОСТАТИКА
- III. Экстраполяция по темпу роста.
- XX съезд КПСС и осуждение культа личности. Экономические реформы конца 50-х – начала 60-х гг. Причины их неудач. Замедление экономического роста.
- А. Электростатическое экранирование
- Аналогия электрического поля в проводящей среде с электростатическим полем
- Атомные электростанции
- Атомные электростанции
- Бальзамы для интенсивного роста волос

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
