Митохондриальные болезни
В 1972 г. Ольсон с соавторами с помощью модифицированной трехцветной окраски обнаружили в поперечнополосатых мышцах волокна с аномально большим количеством митохондрий. Они предложили новый термин – " рваные мышечные волокна ". При электронной микроскопии видно, что митохондрии в таких волокнах увеличены , часто имеют необычную форму и содержат кристаллические включения . Со времени этих исследований представления о митохондриальных миопатиях и наследственных митохондриальных болезнях вообще существенно расширились.
Митохондриям принадлежит ведущая роль в образовании энергии. В результате окисления углеводов, жиров и белков образуются восстановительные эквиваленты, которые переносятся по дыхательной цепи. Высвобождающаяся при этом энергия переходит в энергию электрохимического градиента для протонов на внутренней мембране митохондрий, а та, в свою очередь, используется для синтеза АТФ . Этот процесс - окислительное фосфорилирование . Митохондриальный геном кодирует рРНК и тРНК, участвующие в митохондриальной системе трансляции, и некоторые белки, необходимые для окислительного фосфорилирования. 13 белок-кодирующих митохондриальных генов занимают примерно 77% кодирующих последовательностей мтДНК; на долю двух генов рРНК приходится 14%, а 22 гена тРНК составляют 9% митохондриальной ДНК человека. Многие митохондриальные белки кодируются генами ядерного генома, синтезируются в цитоплазме и затем транспортируются в митохондрии. Поэтому мутации, нарушающие функции митохондрий, могут происходить как в митохондриальном, так и в ядерном геномах.
Дефект любого из ферментов митохондрий нарушает слаженную работу всей "энергетической станции". При этом в первую очередь страдают наиболее энергозависимые ткани и органы - центральная нервная система, скелетные и сердечная мышцы, почки, печень, эндокринные железы. На фоне хронического дефицита энергии в них рано или поздно возникают патологические изменения и развиваются заболевания, которые получили название митохондриальных. Современной медицине известно около 200 таких болезней. В их клинике встречается самая различная патология, но доминируют поражения центральной нервной системы и мышечной ткани. Симптомами, типичными для митохондриальных заболеваний, являются мышечные боли, слабость и атрофия мускулатуры, непереносимость физических нагрузок, птоз, полинейропатия, судороги, отсутствие рефлексов, атрофия зрительного нерва, нейросенсорная тугоухость, мигрени, летаргические состояния, изменения психомоторного развития, олигофрения и деменция.
Мутации, возникшие в митохондриальных генах, передаются в новые митохондрии при делении этих органелл. Получается, что даже в пределах одной клетки присутствуют митохондрии с разными вариантами геномов. Это явление называется гетероплазмией.Человек с мутацией в митохондриальном гене несет смесь нормальной и мутантной ДНК, причем соотношение митохондрий с мутантными и нормальными геномами может быть каким угодно, поэтому выраженность митохондриальных заболеваний у разных больных неодинаковая. В подобных случаях мутации поначалу могут вообще не иметь внешних проявлений. Нормальные митохондрии до поры до времени обеспечивают клетки энергией, компенсируя недостаточность функции митохондрий с дефектами. На практике это проявляется более или менее длительным бессимптомным периодом при многих митохондриальных заболеваниях. Однако рано или поздно наступает момент, когда дефектные формы накапливаются в количестве, достаточном для проявления патологических признаков. Возраст манифестации заболевания варьирует у разных больных. Раннее начало заболевания приводит к более тяжелому течению и неутешительному прогнозу.
Наследование мутаций в митохондриальном геноме носит особый характер. Если гены, заключенные в ядерной ДНК, дети получают поровну от обоих родителей, то митохондриальные гены передаются потомкам только от матери. Это связано с тем, что всю цитоплазму с содержащимися в ней митохондриями потомки получают вместе с яйцеклеткой, в то время как в сперматозоидах цитоплазма практически отсутствует. По этой причине женщина с митохондриальным заболеванием передаёт его всем своим детям, а больной мужчина - нет.
В 1988 году были опубликованы первые работы, в которых показана связь мутаций в мтДНК с рядом заболеваний у человека. Сейчас известно более 110 патогенных точечных мутаций и около 200 делеции, инсерций и других структурных реорганизаций мтДНК человека, индуцирующих развитие наследственных заболеваний.
Проявление мутаций мтДНК тканеспецифично: сердце, мышцы и мозг являются наиболее зависимыми от аномалий процесса окислительного фосфорилирования, с этим и связаны особенности большинства "митохондриальных" синдромов (рис. 11.5).
Каждый из известных сегодня синдромов, вызванных нарушением функционирования митохондрий, определяется какой-либо мутацией следующего типа:
- нуклеотидные замены в генах, кодирующих полипептидные цепи (миссенс-мутации);
- нуклеотидные замены в генах тРНК;
- делеции или вставки в мтДНК;
- мутации, изменяющие число копий мтДНК.
В митохондриальном геноме человека обнаружен ряд единичных делеций размером от 1 до 10 т.п.н. Оказалось, что почти треть пациентов с делециями мтДНК утрачивают один и тот же участок митохондриального генома размером 4977 п.н, несут так называемую обычную делецию. Она фланкирована двумя прямыми повторами размером 13 п.н и расположена между генами atp8 и nad5. Все макроделеции, найденные в мтДНК человека, разделяются на две группы: 1) имеющие короткие прямые повторы в точках разрыва ДНК молекулы; 2) не имеющие таких повторов.
У пациентов, несущих делеции мтДНК, обнаруживаются часто также мтДНК с дупликациями. Описана реорганизация генома, в результате которой объединяются интактная молекула мтДНК - 16,6 т.п.н. с молекулой, несущей делецию - 11,6 т.п.н, при этом образуются кольцевые молекулы размером 28,2 т.п.н.
Патогенность крупных делеции связывают прежде всего с утратой генов сразу нескольких тРНК, что приводит к нарушению трансляции практически всего митохондриального генома. С делецией 4977 п.н. (утрачиваются гены тРНК, расположенные между atp8 и nad5) связаны сразу три синдрома:
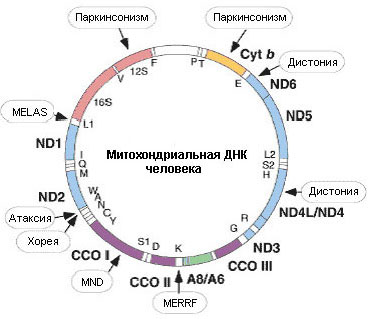
Рисунок. 11.5. Митохондриальный геном человека с указанными позициями делеции 5 т.п.н, а также различных точечных мутаций и связанных с ними синдромов.
- синдром Кернс-Сейра - фатальная мультисистемная патология, проявляющаяся в возрасте 4 - 18 лет пигментным ретинитом, атаксией, атриовентрикулярной блокадой сердца, повышением уровня белка в цереброспинальной жидкости, появлением "рваных" волокон в скелетных мышцах и др.;
- прогрессирующая наружная офтальмоплегия - миопатия, характеризующаяся параличом наружных глазодвигательных мышц;
- синдром Пирсона - гипопластическая анемия, нарушение экзокринной функции поджелудочной железы.
Все три синдрома являются спорадическими, т.е. матери и другие дети тех же родителей остаются здоровыми. Митохондриальные ДНК больных характеризуются гетероплазмией: делетированные молекулы присутствуют в тканях одновременно с нормальными. Причиной заболевания является единичная спонтанная делеция, происходящая рано в оогенезе или эмбриогенезе. До начала дифференциации зародышевых слоев мтДНК не реплицируется. Если мутантная мтДНК по воле случая распределится равномерно между клетками всех зародышевых слоев, разовьется скорее всего мультисистемный синдром Кернс-Сейра; неравномерная сегрегация с накоплением мутантных мтДНК в клетках костного мозга может дать начало синдрому Пирсона, сегрегация исключительно в мышцы приведет к формированию прогрессирующей наружной офтальмоплегии.
Возникнув первоначально в результате мутации, единичная молекула мтДНК с делецией амплифицируется, приводя к появлению триллионов аномальных ДНК у больных с клиническими синдромами. Именно пролиферация дефектных митохондрий (особенно в мышцах) в значительно большей степени, чем присутствие мутантной ДНК в них, приводит к гибели больного.
Множественные митохондриальные делеции были обнаружены в ДНК пациентов с митохондриальной нейрогастроинтестинальной энцефало-миопатией, прогрессивной наружной офтальмоплегией и рядом других симптомов. В этих случаях дефект наследовался по менделевскому типу как аутосомный рецессивный ген, т.е. его первопричина находилась вне митохондриальной ДНК.
Во многих описанных случаях в семьях пациентов, происходящих из самых разных этнических групп, был обнаружен дефектный ядерный ген ти-мидинфосфорилазы (хромосома 22ql3.32-qter). Тимидинфосфорилаза катализирует реакцию превращения тимидина в тимин. У 15 исследованных больных с синдромом MNGIE уровень тимидина в плазме был повышен в 20 и более раз по сравнению с контрольным. Вероятно, нарушенный метаболизм тимидина приводит к нестабильности мтДНК.
Тяжелые клинические последствия имеет процесс почти полной утраты клеткой мтДНК - так называемый синдром мтДНК деплеции. При этом в клетках остается 1 - 30% нормального количества молекул мтДНК. Синдром деплеции также наследуется по аутосомно-рецессивному типу, мутирующий при этом ген пока окончательно не установлен; скорее всего им окажется ядерный ген, кодирующий ДНК-полимеразу гамма. При трансплантации in vitroядер из нормальных клеток в клетки с деплецией митохондриального генома восстанавливается нормальный уровень молекул мтДНК, что доказывает ядерную локализацию данной мутации.
Синдром проявляется в первые недели после рождения; деплеция мтДНК была даже обнаружена в клетках амниотической жидкости плода. Новорожденные страдают множественными нарушениями: фатальная гепатопатия, врожденная или возникающая в первые два года жизни миопатия с генерализованной гипотонией, кардиомиопатией и судорогами (синдром Де Тони-Дебре-Фанкони), атрофией проксимальных групп мышц и утратой сухожильных рефлексов. В тяжелых случаях смерть наступает в первый год жизни; даже при менее массированной деплеции течение болезни быстропрогрессирующее, с летальным исходом в первые три года жизни.
Частота мутаций в генах тРНК в 2 раза выше, чем в белок-кодирующих генах. Подавляющее большинство исследованных до сих пор мутаций митохондриальных генов тРНК являются рецессивными: биохимическая дисфункция проявляется лишь тогда, когда уровень немутантных мтДНК падает ниже 30%.
Известно пять мутаций генов рибосомальной РНК митохондрий, все они вызывают тканеспецифические эффекты. Наиболее часто встречается мутация гена 12S рРНК A1555G. Она находится в высококонсервативной области гена РНК малой субъединицы рибосомы и вызывает врожденную несиндромную потерю слуха. В результате этой мутации изменяется аминогликозид-связывающий сайт 12S рРНК, вследствие чего пациенты становятся чувствительными к находящимся во внешней среде ототоксическим аминогликозидам. Еще четыре мутации генов 12S и 16S рРНК вызывают кардиомиопатию.
У большинства пациентов с синдромом MERRF (миоклонус-эпилепсия, "рваные" красные мышечные волокна, задержка умственного развития, атаксия, атрофия мышц и др.) обнаружена гетероплазматическая транзиция A8344G в гене TPHKLys. Дисфункция дыхательной цепи проявляется при очень высоком уровне мутантных молекул в клетках - не менее 85 - 90%. Матери пациентов с синдромом MERRF несут значительно менее выраженные синдромы либо фенотипически являются здоровыми.
У некоторых пациентов с мутацией A8344G были обнаружены множественные симметричные липомы на шее, которые содержали более высокий уровень мутантной мтДНК, чем окружающие жировые ткани, и, возможно, являлись первым проявлением мутантного фенотипа. Реже встречается вторая мутация гена лизиновой тРНК: транзиция нуклеотида 8356 (Т8356С). Данная мутация также ингибирует белковый синтез в митохондриях и вызывает дефицит дыхательной цепи.
Основная часть обнаруженных мутаций тРНК генов является транзициями и трансверсиями.
Дата добавления: 2022-07-20; просмотров: 230;
Поиск по сайту

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
