Ферментативная кинетика
Ферментативный катализ существенно отличается от неферментативного, в связи с чем в кинетике ферментативных реакций разработаны совершенно особые закономерности. Они позволяют выделить ферментативную кинетику в самостоятельный раздел химической кинетики, в котором изучается зависимость скорости реакций, катализируемых ферментами, от концентрации реагирующих веществ (ферментов и субстратов) и от условий их взаимодействия (температуры, рН, концентрации коферментов и кофакторов, наличия различных эффекторов: активаторов и ингибиторов).
Изучение кинетики ферментативного действия имеет важное теоретическое значение, поскольку только с позиций кинетики можно подойти к решению вопроса о механизме ферментативного действия. Но оно также необходимо с практических позиций, так как только имея определенные сведения о кинетике действия того или иного фермента, можно подобрать оптимальные условия для его работы, а также влиять на его активность в заданном направлении на различных стадиях технологического процесса.
Вопросы, связанные с кинетикой ферментативных реакций, детально изложены в специальных разделах биохимии и энзимологии, поэтому основное внимание уделим тем положениям, которые необходимы для грамотного подхода к работе с ферментами: подбору условий для определения активности фермента, определению начальной скорости ферментативной реакции, выбору субстрата, определению его насыщающей концентрации, оптимуму действия температуры и рН, влиянию кофакторов, активаторов и ингибиторов.
Наличие фермента в растворе или экстракте можно определить исходя из скорости катализируемой им реакции, о которой можно судить либо по накоплению продуктов реакции, либо по убыли субстрата.
В большинстве своем ферментативные реакции являются реакциями смешанного порядка. Типичная кривая хода ферментативной реакции (рис. 8.1) имеет следующий вид:
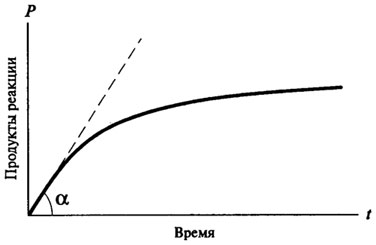
Рис. 8.1.Кривая хода ферментативной реакции во времени
Таким образом, ход ферментативной реакции во времени не может быть описан одним математическим уравнением, поскольку все ферментативные реакции в самом начале своего протекания (когда имеется избыток субстрата и образовалось мало продуктов реакции) являются реакциями нулевого порядка, и только потом они приобретают характер реакции первого или второго порядка. Скорость реакции нулевого порядка со временем не меняется, зависимость количества образовавшегося продукта от времени остается прямо пропорциональной (см. рис. 8.2). Для реакций первого порядка скорость реакции в каждый данный момент времени пропорциональна имеющейся в наличии концентрации субстрата, а следовательно, наблюдается постоянное падение скорости реакции с течением времени (см. рис. 8.3).
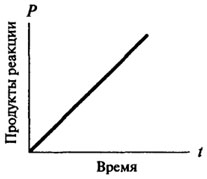
Рис. 8.2.Графическое изображение реакции нулевого порядка
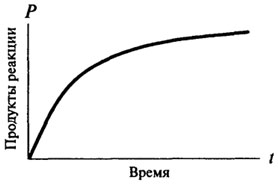
Рис. 8.3.Графическое изображение реакции первого порядка
Для того чтобы правильно определить потенциальные возможности данного фермента как катализатора, нужно учитывать скорость ферментативной реакции в тот момент времени, когда факторы, замедляющие скорость ферментативной реакции (нехватка субстрата, специфическое ингибирование продуктами реакции, частичная тепловая денатурация фермента и др.), не успевают проявить свое действие и наблюдается прямая пропорциональная зависимость между продуктами реакции и временем.
Такая скорость называется начальной скоростью ферментативной реакции и обозначается V0.
На практике V0 определяют графическим методом, для чего строят кривую хода ферментативной реакции во времени. Начальная скорость определяется как тангенс угла наклона касательной, проведенной из начала координат к кривой хода ферментативной реакции (см. рис. 8.1).
V0 = tg a
При работе с конкретным ферментом длительность реакции следует выбирать исходя из экспериментальных данных, по начальной скорости реакции.
В зависимости от задачи, которая стоит перед исследователями или технологами, теми, кто работает с ферментами, выбирается тот или иной подход в этой работе. Имеется в виду следующее.
1. Если необходимо выделить и охарактеризовать фермент из какого-либо биологического объекта, пищевого сырья, следует применить либо известные схемы выделения и очистки, или разработать оптимальную схему для данного фермента, варьируя и испытывая различные сочетания основных этапов очистки и выделения ферментов (белков): экстракцию, различные режимы осаждения, гель-хроматографию и другие методы, основанные на различиях в физико-химических характеристиках отдельных ферментов (см. также гл. 2). При этом на каждом этапе выделения и очистки следует характеризовать ферментный препарат по ферментативной активности и содержанию белка. В этом случае определение ферментативной активности (определение F0) проводят с использованием стандартного субстрата; выявляют оптимальные значения рН и температуры. И все дальнейшие исследования проводят при насыщающей концентрации субстрата, оптимуме температуры и рН. Изучение влияния специфических активаторов и ингибиторов позволяет в этом случае получить ценные сведения о строении активного центра и возможном механизме каталитического действия. Здесь необходимо подчеркнуть важность тщательного методического подхода при работе с ферментами. Не следует жалеть времени и усилий на выбор режима экстракции (продолжительность, температура, экстрагент, тип экстракции - исчерпывающая или нет), выбор методики определения активности, ее отработку и возможную модификацию для данного конкретного объекта исследования; кроме того, работа с ферментами различной степени очистки также имеет свои особенности, свою специфику: они обладают разной рН- и термостабильностью и, помимо этого, могут по-разному реагировать на воздействие различных факторов.
2. Если задача заключается в определении того, каким образом будет вести себя данный фермент (ферментный препарат) в конкретном режиме рассматриваемой пищевой технологии, необходимо проводить исследование ферментативного действия при условиях данного технологического процесса (концентрация субстрата, длительность, рН, температура, влажность), изучить влияние различных компонентов пищевого сырья и используемых добавок на активность фермента с целью определить возможность и способы влияния на ферментативный процесс в желаемом направлении.
Перейдем к рассмотрению факторов, влияющих на скорость ферментативных реакций.
Влияние концентрации субстрата на скорость ферментативной реакции.Концентрация субстрата является важнейшим фактором, определяющим скорость ферментативной реакции. Еще в 1902г. В. Анри при изучении реакции ферментативного гидролиза сахарозы предположил, что фермент р-фруктофуранозидаза взаимодействует со своим субстратом, затем это соединение распадается, фермент остается в первоначальном виде, а субстрат сахароза оказывается расщепленной на глюкозу и фруктозу.
Это предложение было в дальнейшем развито Л. Михаэлисом и M. Ментен. В 1913 г. они постулировали следующие уравнения ферментативной реакции:
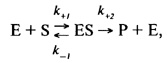
где k+1- константа скорости реакции образования комплекса ES, k-1 k+2 - константы скорости реакции распада комплекса ES в двух направлениях.
Тогда Ks- константа диссоциации комплекса ES равна отношению констант скоростей обратной и прямой реакции:
Ks=
| k-1 |
| k+1 |
Исходя из закона действующих масс, можно записать следующее уравнение:
[S] ·([E0] -[ES]) = Ks ·[ES],
где [E0] - концентрация фермента в начале ферментативной реакции, [S] - концентрация субстрата, (ES] - концентрация комплекса "фермент-субстрат", [E0]-[ES] - концентрация фермента, не связанного в комплексе с субстратом.
В ходе ферментативной реакции в любой момент времени фермент существует в двух формах: свободной и связанной, т. е. в форме комплекса ES.
Скорость ферментативной реакции будет максимальной при такой концентрации субстрата, когда весь фермент перейдет в комплекс ES, т.е. когда все активные центры насыщены субстратом и дальнейшее увеличение концентрации субстрата не приведет к увеличению скорости реакции.
Преобразуя представленное выше уравнение, получим выражение, которое будет иметь следующий вид:
V0=
| Vmax[S] |
| K s+[S] |
Это уравнение названо уравнением Михаэлиса-Ментен. Оно имеет огромное значение для выражения зависимости действия ферментов от концентрации субстрата. Однако оно содержит и ряд недостатков, в частности, при его выводе было сделано несколько допущений, например, не учитывалась вторая стадия ферментативной реакции - образование E и P. В связи с этим был предложен рад усовершенствованных уравнений, с учетом влияния образовавшихся продуктов реакции. В настоящее время наиболее широко используют уравнение Холдейна-Бриггса. Оно имеет следующий вид:
V0=
| Vmax[S] |
| K m+[S] |
В этом уравнении вместо K s - константы диссоциации комплекса ES, который присутствует в уравнении Михаэлиса-Ментен, стоит Km-константа Михаэлиса (в числителе которой находятся константы скоростей реакций, ведущих к распаду комплекса ES в двух направлениях):
Km=
| k-1+K+2 |
| K +1 |
Поскольку K s =
| k-1 |
| k+1 |
,то Km = Ks+
| k+2 |
| k+1 |
, то есть Km всегда больше Ks.
Для того, чтобы графическая зависимость, выражающая влияние концентрации субстрата на начальную скорость ферментативной реакции, из гиперболической преобразовалась в прямолинейную, что, очевидно, представляет большее удобство в экспериментальной практике, уравнение Холдейна-Бриггса было преобразовано Лайнуивером и Берком по методу двойных обратных величин.
| V0 |
=
| K m |
| Vmax |
·
| [S] |
+
| Vmax |
Графически это выгладит так:
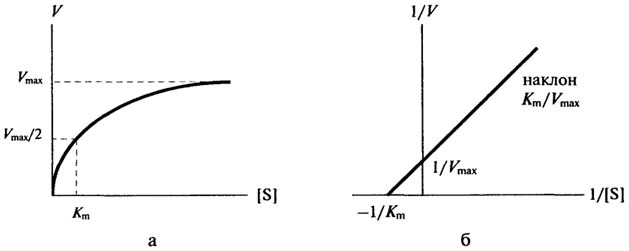
Рис. 8.4.Влияние концентрации субстрата на начальную скорость ферментативной реакции (а - по методу Михаэлиса-Ментен, б - по методу Лайнуивера-Берка)
Величина Km - это ключевой кинетический параметр; если [S] = K m, то V= Vmax/2, следовательно, константа Михаэлиса численно равна концентрации субстрата (в молях на литр), при которой скорость реакции равна половине максимальной.
Приблизительное значение Km можно получить простым графическим способом, как это показано на рис. 8.4 а; однако в этом способе достаточно велика погрешность в нахождении Vmax. Значительно удобнее пользоваться прямолинейной зависимостью при обработке данных по методу двойных обратных величин, рис. 8.4 б. В этом случае можно получить более точное значение Km.
Таблица 8.1.Значение констант Михаэлиса - К (мМ/л) для некоторых ферментов
| Фермент | Субстрат | К |
| Каталаза | H2 O 2 | 25,0 |
| Гексокиназа | АТФ | 0,4 |
| Глюкоза | 0,05 | |
| Фруктоза | 1,5 | |
| Химотрипсин | Глицил-тирозинил-глицин | 10,8 |
| N-бензол-тирозин-амид | 2,5 | |
| P - Галактозидаза | Лактоза | 4,0 |
Источником множества недоразумений как в прошлом, так и в настоящем, является некорректное использование термина "константа Михаэлиса" и двух символов Ks и Km для обозначения величин отнюдь неидентичных, несмотря на совершенно четкие рекомендации Комиссии по ферментам Международного Биохимического Союза. Первая величина -Ks- константа равновесия, выражаемая отношением Ks =
| k-1 |
| k+1 |
,
характеризует сродство фермента к субстрату (или, иначе, прочность комплекса ES), причем существует обратная пропорциональность между величиной Ks и сродством фермента к субстрату. Вторая величина -Km-соответствует концентрации субстрата, при которой V= Vmax/ 2. Часто свойство Ks ошибочно приписывают Km. Ha самом деле Km будет являться мерой сродства фермента к субстрату только в том единственном случае, когда величина k+2 будет настолько мала, что Km практически совпадет с Ks.
Многие ферменты катализируют реакции с участием двух субстратов. К так называемым бимолекулярным реакциям относятся реакции переноса химических группировок с одного соединения на другое, реакции синтеза, окислительно-восстановительные реакции. Такие реакции могут протекать по двум различным механизмам. В реакциях первого типа, называемых реакциями единичного замещения, два субстрата А и В образуют с ферментом комплекс EAB, который затем распадается с образованием продуктов реакции С и Д. Второй тип двухсубстратных реакций протекает по механизму двойного замещения (механизм типа "пинг-понг"). В этих реакциях с активным центром фермента в каждый момент времени связан только один из двух субстратов.
При исследовании кинетики бимолекулярных реакций концентрацию одного из субстратов оставляют постоянной (В), а второго - изменяют (А). В этом случае в координатах 1/V от 1/[A] можно получить "кажущееся" значение К т. Истинное значение Vmax и К Bmполучают при исследовании нескольких концентраций субстрата В. Точно так же поступают при определении K Am(когда концентрация А постоянна, а концентрация В варьируется). Кт по отношению к различным субстратам в одной и той же реакции могут быть различными - это хорошо видно из следующего примера.
Реакция катализируемая алкогольдегидрогеназой:
CH3CH2OH + НАД+ ↔ CH3COH + НАДН + H+
этанол уксусный альдегид
Значение Кт для алкогольдегидрогеназы дрожжей:
| Субстрат | Кт, М/л |
| НАД+ | 1,0 ×10-4 |
| CH3CH2OH | 2,4 ×10-2 |
| НАДН + H+ | 3,5 ×10-5 |
| CH3COH | 1,0 ×10-4 |
Дата добавления: 2016-12-16; просмотров: 2854;
Поиск по сайту
Узнать еще
- II.1.4 ФИЗИЧЕСКАЯ КИНЕТИКА
- III.1.4 ФИЗИЧЕСКАЯ КИНЕТИКА
- Биологическая кинетика
- Кинетика абсорбции.
- Кинетика гетерогенных высокотемпературных процессов и методы выявления их ведущего звена
- Кинетика двусторонних (обратимых) реакций
- Кинетика десульфурации металла за счет твердых тел
- Кинетика диффузионных процессов.

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
