Делокализация заряда энергетически выгодна.
Таким образом, увеличение числа алкильных заместителей у катионного центра стабилизирует катион, понижает его энергию. Наиболее стабильны третичные карбокатионы, менее стабильны вторичные, затем первичные, и совсем уж неустойчив метилкатион:
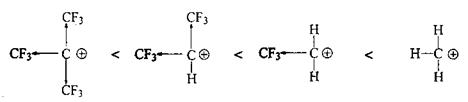
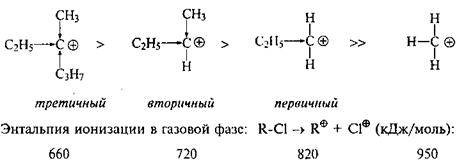 Эти цифры наглядно показывают, насколько различаются затраты энергии необходимые для образования различных катионов.
Эти цифры наглядно показывают, насколько различаются затраты энергии необходимые для образования различных катионов.
Трифторметильные группы вследствие своей высокой электроотрицательности оттягивают электронную плотность вдоль σ-связи и тем самым увеличивают, концентрируют положительный заряд на катионном центре, что приводит к дестабилизации катиона:
К аналогичной индуктивной дестабилизации приводит накопление электроотрицательных атомов галогена при катионном центре:
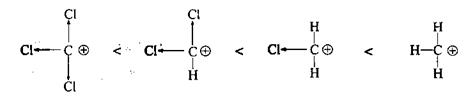
[за счет мезомерного эффекта галогены могут оказывать противоположное действие – стабилизировать карбокатион; см. далее.]
Следующие примеры показывают, что от стабильности карбокатионных интермедиатов может зависеть направление реакции
2-Метилпропен в реакции с НСl может образовать карбокатионы с тремя (третичный ион) или с одним алкильным заместителем (первичный ион). Поскольку единственным продуктом реакции является третичный хлорид, 2-метил-2-хлорпропан, образование третичного карбокатиона, очевидно, предпочтительно.
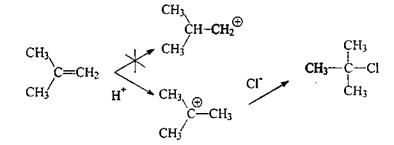
В ходе взаимодействия НХ с алкенами часто происходят структурные перегруппировки. Так, реакция 3,3-диметил-1-бутена с НСl приводит к смеси равных количеств ожидаемого 3,3-диметил-2-хлорбутана и продукта перегруппировки - 2,3-диметил-2-хлорбутана.
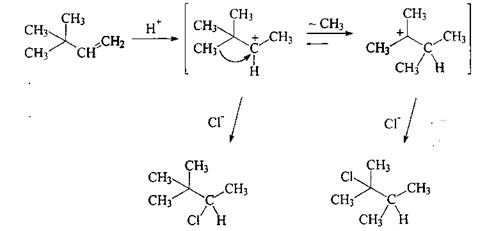 В данном случае вторичный карбокатион перегруппировывается в более стабильный третичный путем перемещения метильной группы вместе с парой электронов σ-связи. Изменяется направление реакции.
В данном случае вторичный карбокатион перегруппировывается в более стабильный третичный путем перемещения метильной группы вместе с парой электронов σ-связи. Изменяется направление реакции.
Кислотность спиртов и карбоновых кислот. Согласно определению Бренстеда, кислоты представляют собой вещества, отщепляющие протоны, а основания - вещества, присоединяющие протоны (протонная теория кислот и оснований).
НА + Н2O D A–+Н3O+
Силу кислоты характеризуют константой диссоциации (Ка):
Ка = [А–][Н3O+]/[НА] pKa = –lgKa
Соединения с малыми значениями Ка (большими рКа) являются слабыми кислотами, соединения с большими значениями Ка (малыми рКа) - сильными кислотами. Таким образом, кислотность есть термодинамическая характеристика: она определяется положением равновесия диссоциации, т.е. энергией исходного и конечного состояний равновесной системы.
Кислотность соединения Н-А зависит:
1) от полярности связи Н-А, т.е. от степени смешения электронов от атома водорода Н→А, "подготовленности" Н к отщеплению в виде протона (Н+);
2) от стабильности образующегося аниона А–.
Эту зависимость иллюстрируют свойства следующих простых соединений, кислотность которых последовательно увеличивается от метана до муравьиной кислоты:
СН4 D СН3– + Н+ рКа ~ 43
СН3ОН D СН3O– + Н+ рКа = 15,54
С6Н5ОН D С6Н5O– + Н+ рКа = 9,95
НСООН D НСОО– + Н+ рКа = 3,77.
| Спирт | рКа |
| (СН3)3СОН | 18,00 |
| CH3CH2OH | 16,00 |
| HOH | 15,74 |
| CH3OH | 15,54 |
| CF3CH2OH | 12,43 |
| (CF3)3COH | 5,4 |
| HCl | -7,00 |
Спиртыпо кислотности близки к воде (слабые кислоты), причем значительную роль в способности диссоциировать играет их структура. В галогенпроизводных спиртов благодаря отрицательному индуктивному эффекту (–I) галогенсодержащие заместители делокализуют отрицательный заряд, тем самым стабилизируют анион А–, смещают равновесие в сторону его образования, а значит, увеличивают кислотность. В некоторых случаях это увеличение значительно:
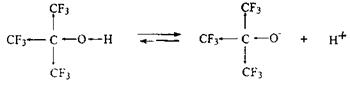
Электронодонорные алкильные группы (+I-эффект), напротив, повышают электронную плотность на атоме кислорода, тем самым дестабилизируют анион и, следовательно, понижают кислотность.
[Кроме того, объемистые алкильные группы мешают сольватации аниона водой, что еще больше его дестабилизирует.]
Аналогичные закономерности наблюдаются для карбоновых кислот.Электроноакцепторные группы, соединенные с карбоксилом, индуктивно оттягивают электронную плотность, тем самым делокализуя отрицательный заряд, стабилизируя карбоксилат-анион и увеличивая кислотность:
Электронодонорные группы оказывают прямо противоположный эффект, концентрируя заряд, дестабилизируя карбоксилат-анион и уменьшая кислотность. Эти закономерности хорошо прослеживаются в приведенных ниже сериях кислот:
| Кислота | рКа | Кислота | рКа |
| (СН3)3ССООН | 5,05 | СН3СН2СН2СООН | 4,82 |
| СН3СН2СООН | 4,88 | СlСH2СН2СН2СООН | 4,52 |
| CH3COOH | 4,76 | СН3СНСlСН2СООН | 4,05 |
| HCOOH | 3,77 | СН3СН2СHСlСООН | 2,86 |
| СН3СООН | 4,76 | CH3COOH | 4,76 |
| IСН2СООН | 3,16 | СlСН2СООН | 2,86 |
| ВrСН2СООН | 2,90 | Сl2СНСООН | 1,25 |
| СlСН2СООН | 2,86 | Сl3ССООН | 0,65 |
| FCH2COOH | 2,57 | ||
| СН3ОСН2СООН | 3,53 | (CH3)3N 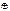 CH2COOH CH2COOH
| 1,83 |
| N≡ССН2СООН | 2,47 | O2NCH2COOH | 1,68 |
Хлорбутановые кислоты (правый верхний угол таблицы) показывают, как быстро уменьшается действие электроотрицательного заместителя при удалении от карбоксила (затухание -I-эффекта): 4-хлорбутановая кислота по своей кислотности практически не отличается от самой бутановой кислоты.
Ненасыщенные карбоновые кислоты сильнее своих насыщенных аналогов - сказывается слабый -I-эффект непредельных групп, обусловленный увеличением s-характера атома углерода:
| СН3СН2СООН | CH2=CHCOOH | СН≡ССООН |
| рКа 4,88 | 4,25 | 1,84 |
Аналогично действуют ароматические заместители. Так, бензойная кислом сильнее циклогексанкарбоновой:
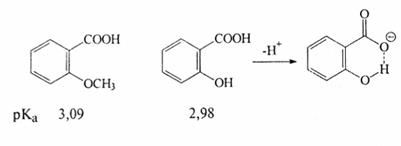
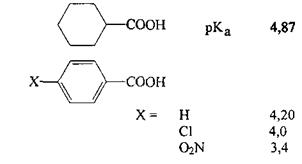 Удаленные пара-заместители в бензольном кольце оказывают довольно слабое воздействие на стабильность аниона (затухание -I-эффекта). орто-Заместители действуют сильнее. Кроме того, они могут дополнительно стабилизировать анион, например, за счет внутримолекулярной водородной связи:
Удаленные пара-заместители в бензольном кольце оказывают довольно слабое воздействие на стабильность аниона (затухание -I-эффекта). орто-Заместители действуют сильнее. Кроме того, они могут дополнительно стабилизировать анион, например, за счет внутримолекулярной водородной связи:
Основность- это есть способность присоединять протон, количественно оценивается с помощью величин констант равновесия Кb для взаимодействия основания с водой:
В: + Н2O — НВ+ + НО-
Кb = [НВ+][НО-]/[В]
Показателем силы основания является также кислотность сопряженной кислоты НВ+ (Ка или рКа): чем сильнее эта кислота, тем слабее сопряженное основание В.
Основность должна увеличиваться с увеличением плотности отрицательного заряда на атоме. Поэтому сила оснований тем больше, чем сильнее электронодонорные влияния, повышающие электронную плотность (+I-эффект). И наоборот, основность тем меньше, чем сильнее электроноакцепторность (-I-эффект) заместителей. Например, анионы спиртов и карбоновых кислот можно расположить в следующие ряды по уменьшению их основности (см. в предыдущем разделе значения рКа):
(CH3)3CO– > (CH3)2CHO– > CH3CH2O– > CH3O– > HO– > C6H5O– > CH3COO– > CCl3COO–.
С6H5O– > п-ClC6H4O– > п-O2NC6H4O–
Важнейшими органическими основаниями являются амины. Они значительно более сильные основания, чем спирты, эфиры или вода (но не анионы этих соединений !). В водном растворе амина вода действует как протонная кислота, отдавая протон амину:
R3N + Н2O D R3NH+ + ОН–.
Измерения основности аминов показывают, что замена атомов Н в аммиаке на более электронодонорные алкильные заместители увеличивает основность, но не сильно. Закономерность нарушается для третичных аминов они слабее как основании, чем вторичные Причина этого явления не связана с электронными эффектами заместителей при атоме азота: три алкильные группы затрудняют гидратацию образующегося катиона, что делает его менее стабильным. В газовой фазе третичные амины, как и ожидалось, наиболее основны.
| Амин | Kb | pKa (R3NH+) |
| NH3 | 1,8·10-5 | 9,26 |
| Первичные алкиламины: CH3NH2 CH3CH2NH2 | 5,1·10-4 | 10,64 10,75 |
| Вторичные алкиламины: (CH3)2NH (CH3CH2)2NH | 10,0·10-4 | 10,73 10,94 |
| Третичные алкиламины: (CH3)3N (CH3CH2)3N | 5,6·10-4 | 9,79 10,75 |
Ароматические амины менее основны, чем алкиламины и аммиак из-за уменьшения электронной плотности на атоме азота вследствие отрицательного индуктивного и, главным образом, мезомерного (см. далее) эффектов ароматической группы.
Мезомерный эффект
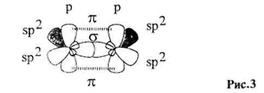 Так называемая "двойная" связьмежду двумя sp2-гибридизован-ными атомами (С=С, С=O, C=N и т.п.) представляет собой сочетание σ-связи (см. стр. 2) и π-связи. Ковалентная π-связь образуется обшей для двух соединенных атомов парой р-электронов, причем занимаемые ими гантелеобразные орбитали параллельны и перекрываются боковыми сторонами.
Так называемая "двойная" связьмежду двумя sp2-гибридизован-ными атомами (С=С, С=O, C=N и т.п.) представляет собой сочетание σ-связи (см. стр. 2) и π-связи. Ковалентная π-связь образуется обшей для двух соединенных атомов парой р-электронов, причем занимаемые ими гантелеобразные орбитали параллельны и перекрываются боковыми сторонами.
В отличие от σ-связи, максимальная электронная плотность π-связи расположена не на прямой между ядрами атомов, а сбоку, по разные стороны от нее. Такое перекрывание менее эффективно, поэтому энергия (прочность) двойной связи значительно меньше, чем удвоенная энергия простой (σ) связи: Е(С-С) = 350 кДж/моль, Е(С=С) = 600 кДж/моль (а не 2 • 350 = 700 кДж/моль).
Если две двойные связи находятся рядом, происходит перекрывание их π-систем (аналогично перекрыванию p-орбиталей, Рис.3), и образуется общее для четырех атомов электронное π-облако. При этом энергия системы понижается, молекула становится более устойчивой, чем в случае невзаимодействующих двойных связей (для системы связей С=С-С=С энергетический выигрыш составляет около 16 кДж/моль). Это явление называется сопряжением.Поскольку речь идет о перекрывании орбиталей π-электронов, его следует классифицировать как π-π-сопряжение.
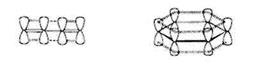
Сопряжение π-связей, разделенных одной σ-связью, приводит к усреднению их параметров: центральная простая связь приобретает частичный "двойной" характер, становится прочнее и короче, а двойные - несколько ослабевают и удлиняются. Особый случай – сопряжение трех двойных связей, находящихся в одном шестичленном цикле, которое является причиной резкого увеличения термодинамической и химической устойчивости (ароматичность). В ароматических молекулах усреднение доходит до предела: все связи С-С в бензоле одинаковы и имеют характеристики средние между простыми и двойными. Энергетический выигрыш от сопряжения для бензола составляет 152 кДж/моль.
Для описания подобных явлений была предложена концепция резонанса или мезомерии, в которой реальное строение молекулы рассматривалось как наложение, резонанс нескольких не существующих в действительности предельных структур (мезомерия = "между частями"). Несмотря на их гипотетичность, только учет свойств всех этих структур  позволяет отразить действительное состояние молекулы ("гибрид"). Эту взаимосвязь предельных формул показывают с помощью двойной стрелки которую ни в коем
позволяет отразить действительное состояние молекулы ("гибрид"). Эту взаимосвязь предельных формул показывают с помощью двойной стрелки которую ни в коем  случае нельзя смешивать со знаком равновесия
случае нельзя смешивать со знаком равновесия
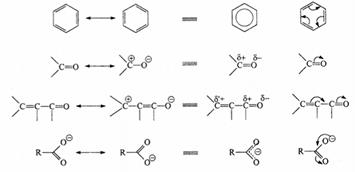
[Согласно методу резонанса,гипотетические резонансные структуры считаются вносящими вклад в реальную структуру молекулы, энергия которой меньше, чем энергия любой из вносящих вклад структур. Несмотря на искусственность в значительной степени устаревшей концепции резонанса, она чрезвычайно полезна при качественном рассмотрении и обобщении фактического материала органической химии. Применяя метод резонанса следует придерживаться следующих основных правил:
1) Все резонансные структуры должны содержать одинаковое число спаренных электронов и иметь идентичное расположение атомов в пространстве, совпадающее с геометрией реальной молекулы.
2) При наличии элементов, сильно различающихся по электроотрицательности, следует учитывать тзкже ионные структуры.
3) Вклад резонансной структуры в "гибрид" тем больше, чем ниже ее энергия.
4) Резонансная стабилизация будет наибольшей, когда есть две или больше
эквивалентные структуры, имеющие наиболее низкую энергию (как у бензола).]
В сопряжении с кратной связью могут находиться не только другие кратные связи, но и неподеленные ("свободные") пары электронов (n-π-сопряжение):

а также катионные или анионные центры, т.е. пустые р-орбитали, или орбитали, несушие избыточный отрицательный заряд (р-π-сопряжение):
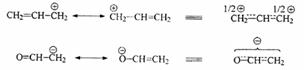
Кратные связи между атомами, имеющими разную электроотрицательность, поляризованы так же, как и простые. Поляризация σ-связей происходит по механизму индуктивного эффекта (I), который передается по цепи, но быстро затухает - через 3-4 связи. Поляризация системы π-связей происходит аналогично. Однако, благодаря большему объему, подвижности, деформируемости π-облаков (т.е. большей поляризуемости) смешение электронов к электроотрицательному атому оказывается значительно сильнее, чем в случае σ-связей, и передается по цепи сопряженных кратных связей, неподеленных электронныхпар и р-орбиталей на гораздо большее расстояние без существенного ослабления.
Смещение электронной плотности π-связей (р-орбиталей) вследствие различия электроотрицательностей атомов или групп называется
Дата добавления: 2018-11-26; просмотров: 1674;
Поиск по сайту
Узнать еще
- I блок — энергетический.
- А. Метод заряда с измерением характеристик электрического поля
- Б. Метод заряда с измерением характеристик магнитного поля
- Баланс мощностей и энергетические характеристики электропривода
- Балансы электроэнергии и топливно-энергетических ресурсов, их состояние и анализ
- Биоэнергетические основы ЭКГ.
- Биоэнергетический анализ.
- Бортовые энергетические системы летательных аппаратов

Публикации по технике и механике

Публикации по биологии

Публикации по информатике

Публикации по строительству

Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по географии

Публикации по медицине
