Кинетические доказательства
Скорость гомогенной реакции определяется скоростью расходования реагентов или скоростью появления продуктов. Почти всегда скорость реакции меняется во времени, так как обычно она пропорциональна концентрации, а концентрация реагентов со временем уменьшается. Однако не всегда скорость реакции пропорциональна концентрации всех реагентов. В некоторых случаях изменение концентрации какого-либо реагента не оказывает никакого влияния на скорость, в то же время в других случаях скорость может быть пропорциональна концентрации вещества (например, катализатора), которое даже не фигурирует в стехиометрнческом уравнении реакции. Изучение вопроса о том, какие именно реагенты оказывают влияние на скорость, может очень много сказать о механизме реакции,
Если скорость пропорциональна изменению концентрации только одного реагента, А, закон скорости (скорость изменения концентрации А за время г] имеет вид
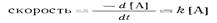
тде коэффициент К— константа скорости реакции. Знак минус означает, что концентрация А уменьшается во времени. Реакции, которые следуют приведенному закону скорости, называют реакциями первого порядка. Единицей измерения К в реакциях первого порядка служит с-1. Скорость реакции второго порядка пропорциональна концентрации двух реагентов или квадрату концентрации одного реагента:
Единицей измерения К в реакции второго порядка является л/ моль • с или другие единицы, выражающие обратную величину концентрации или давления в единичный интервал времени.
Аналогичное выражение можно написать и для реакций третьего порядка. Если скорость реакции пропорциональна [А] и [В], говорят, что реакция имеет первый порядок по А и по В и второй общий порядок. Скорость реакции можно измерять либо по реагенту, либо по продукту, но определенные таким образом скорости необязательно будут одинаковы
Закон скорости реакции — это экспериментально устанавливаемый факт. Из него пытаются узнать молекулярность реакции, которую можно определить как число молекул, объединяющихся для образования активированного комплекса. Если удается узнать, сколько молекул и какие именно участвуют в образовании активированного комплекса, это уже много говорит о механизме реакции. Экспериментально определяемый порядок реакции необязательно совпадает с молекулярностью. Любая реакция независимо от того, сколько стадий она включает, подчиняется только одному закону скорости, но каждая стадия механизма имеет свою собственную молекулярность. Для одностадийных реакций (идущих без образования интермедиатов) порядок —это то же самое, что молекулярность. Одностадийная реакция первого порядка всегда мономолекулярная; одностадийная реакция второго порядка по А всегда включает две молекулы А; если реакция имеет первый порядок по А и по В, молекула А реагирует с одной молекулой В и т. д. Если реакция происходит в две или несколько стадий, порядок каждой стадии — это то же самое, что молекулярность той же стадии. Это позволяет прогнозировать закон скорости для любого предполагаемого механизма, хотя такие расчеты могут оказаться трудоемкими. Если одна из стадий механизма значительно медленнее всех остальных, что встречается очень часто, то скорость реакции по существу будет такой же, как скорость самой медленной стадии, которую поэтому называют определяющей скорость, или лимитирующей стадией.
Все многостадийные реакции можно разделить на две большие группы.
1, Первая стадия медленнее, чем все последующие, н потому является лимитирующей. В таких случаях закон скорости включает реагенты, участвующие в медленной стадии.
2. Если первая стадия не является лимитирующей, вывести закон скорости намного сложнее.
В лимитирующей стадии реакции могут участвовать реагенты А и В, хотя в уравнении скорости появляется только [А]. Это происходит при наличии большого избытка В, скажем, в 100 раз превышающего А по молярному объему, При этом па реакцию с А расходуется только 1 моль В, а 99 молей В остаются неизрасходованными, В таких случаях очень трудно измерить изменение концентрации В во времени, и это редко пытаются делать, особенно если В одновременно является растворителем. Поскольку концентрация избыточного реагента В практически не меняется во времени, реакция имеет кажущийся первый порядок по А, хотя в действительности и А и В участвуют в лимитирующей стадии, Такие реакции часто называют реакциями псевдопервого порядка. Псевдопорядок реакции возникает также в тех случаях, когда одним из реагентов является катализатор, концентрация которого не меняется во времени, так как он регенерируется так же быстро, как и расходуется, или когда реакцию проводят в среде, где поддерживается постоянная концентрация реагента. Условия псевдопервого порядка часто используются в кинетических исследованиях для удобства проведения экспериментов и расчетов.
Реальному измерению поддается изменение во времени концентрации продукта или реагента, присутствующего не в из-быточной концентрации. Для таких измерений существует множество методов, и выбор метода зависит от его удобства применительно к изучаемой реакции, Упомянем наиболее распространенные методы
1. Периодическая или непрерывная запись спектра, Во многих случаях реакцию можно провести в кювете, помещенной в прибор, и тогда задача сводится к периодическому или непрерывному считыванию показаний прибора, Для этих целей широко используются поляриметрия, ИК-, УФ-, ЯМР- и ЭПР-спектроскопия [24].
2. Остановка реакции и анализ реакционной смеси. Проводят серию реакций и каждую останавливают через разные промежутки времени (например, резким понижением температуры или добавлением ингибитора). Реакционную смесь анализируют, используя титрование, газовую хроматографию, поляриметрию, спектральные и другие методы.
3. Взятие аликвотных проб через определенные интервалы времени. Каждую аликвотную пробу анализируют известными методами.
4. Для газофазных реакций измеряют изменения общего давления.
5. Калориметрические методы. Через определенные интервалы времени измеряют количество выделившегося или поглощенного тепла.
Для кинетических измерений очень быстрых реакций существуют специальные методы. В результате таких измерений обычно получают график, показывающий изменение концентрации во времени. Чтобы вывести закон скорости и определить значение К, полученные данные нужно правильно интерпретировать.
При изучении скоростей реакции часто получаются линейные графики, которые несложно интерпретировать, однако во многих случаях результаты не столь просты. Так, иногда реакция может иметь первый порядок при низких концентрациях, но второй при высоких. Иногда получают дробный или даже отрицательный порядок реакции, Интерпретация сложных кинетических данных требует большого искусства и значительных усилий. Даже относительно простая кинетика часто вызывает проблемы при интерпретации данных, связанные с трудностью проведения достаточно точных измерений.
Кинетическую информацию можно получить с помощью одного из специальных методов ЯМР-спектроскопии, основанного на изучении формы линий. Такая возможность связана с тем, что спектр ЯМР имеет собственный временной фактор: если протон меняет свое окружение медленнее чем 103 см, в спектре наблюдаются отдельные сигналы для каждого положения протона. Например, если скорость вращения вокруг связи С—N в N,N-диметилацетамиде не превышает 103 оборотов в секунду, две N-метильные группы будут иметь разные химические сдвиги, так как они неэквивалентны: одна из них находится в цис-, а другая в транс-положении к атому кислорода. Однако если окружение метильных протонов меняется со скоростью, превышающей 103 с-1, в спектре появится только один сигнал с химическим сдвигом, усредненным между двумя индивидуальными положениями. Во многих случаях при низких температурах наблюдаются раздельные сигналы, которые при повышении температуры сливаются, так как при этом скорость взаимопревращений увеличивается, преодолевая рубеж 103 1/с, Следя за изменением формы линий в зависимости от температуры, очень часто можно рассчитать скорость реакций или конформационных превращений [30]. Метод применим к форме сигналов не только протонов, но и других атомов, дающих сигналы в ЯМР-спектрах, а также к спектрам ЭПР.
Из кинетических данных можно получить различного типа информацию о механизмах реакций,
1. По порядку реакции можно судить о том, какие молекулы и в каком количестве участвуют в лимитирующей стадии. Такая информация очень полезна и часто оказывается решающей для выяснения механизма реакции. Для любого механизма, постулируемого для данной реакции, можно вывести соответствующий закон скорости, пользуясь описанными выше методами. Если полученный экспериментально закон с ним не согласуется, значит предполагаемый механизм ошибочен. Однако часто бывает трудно связать порядок реакции с ее механизмом, особенно когда порядок дробный или отрицательный. Кроме, того, часто встречаются случаи, когда два пли несколько предполагаемых механизмов кинетически неразличимы, так как им соответствует один и тот же закон скорости.
2. По-видимому, самыми полезными кинетическими данными являются сами константы скорости, поскольку по ним можно судить о влиянии на скорость реакции таких факторов, как строение реагентов, природа растворителя, ионная сила, катализатор и др.
3. При измерении скорости при разных температурах в большинстве случаев получают линейный график зависимости 1пК от 1/Т (где Т — абсолютная температура) с отрицательным наклоном. По этому графику можно рассчитать Еа — аррениусовскую энергию активации реакции.
Изотопная метка
Замена водорода в молекуле реагента атомом дейтерия часто приводит к изменению скорости реакции. Такие изменения известны как изотопные эффекты дейтерия, которые выражаются отношением KH/KD. В основном состоянии колебательная энергия связи (называемая энергией нулевых колебаний) зависит от массы атомов и при увеличении приведенной массы понижается. Поэтому связи D—С, D—О, D—N и др. в основном состоянии имеют более низкую энергию, чем соответствующие связи Н—С, Н—О, Н—N и др. Следовательно, полная диссоциация связи в дейтерированном соединении требует больше энергии, чем в соответствующем изотопно незамещенном соединении. Если связи Н—С, Н—О или Н—N вообще не разрываются в ходе реакции или разрываются не в лимитирующей стадии, замещение водорода дейтерием практически не оказывает никакого влияния на скорость (об исключениях будет сказано ниже), но если эти связи разрываются в лимитирующей стадии, то скорость при замещении дейтерием понижается.
Изотопные эффекты представляют ценный диагностический инструмент для определения механизмов реакций, Например, тот факт, что скорость бромирования ацетона не зависит от концентрации брома, приводит к предположению о том, что лимитирующей стадией реакции является таутомеризация ацетона. В свою очередь лимитирующая стадия таутомеризации заключается в разрыве связи С—Н. Поэтому при бромироваиии дейтерированного ацетона должен наблюдаться значительный изотопный эффект. Действительно, было найдено, что величина КН/КD составляет около 7. Обычно изотопные эффекты дейтерия меняются от 1 (отсутствие эффекта) до 7—8, однако в некоторых случаях наблюдались и гораздо большие значения. Отношения КН/КD с величинами меньше 1 называют обратными изотопными эффектами. Максимальные изотопные эффекты наблюдаются в тех случаях, когда в переходном состоянии водород симметрично связан с атомами, между которыми осуществляется его перенос, При замещении водорода тритием величина изотопных эффектов превышает соответствующие эффекты дейтерия.
Изотопные эффекты дейтерия наблюдаются иногда даже в тех случаях, когда в реакции вообще не происходит разрыва связи С—Н. Такие эффекты называют вторичными изотопными эффектами; соответственно рассмотренные ранее эффекты относят к типу первичных изотопных эффектов. Вторичные изотопные эффекты можно разделить на α- и β-эффекты в соответствии с положением дейтерия по отношению к разрывающейся связи. Природа изотопных эффектов β-дейтерия была предметом многих дискуссий; наиболее вероятным представляется объяснение с точки зрения гиперконъюгацнн в переходном состоянии. Эти эффекты возрастают, когда переходное состояние имеет значительный карбокатиониый характер. Хотя рассматриваемая связь С—Н не разрывается в переходном состоянии, карбонатной стабилизируется вследствие гнперконъюгацпп, в которой эта связь участвует. Благодаря пшерконъюгации разность колебательной энергии между связями С—Н и С—В в переходном состоянии меньше, чем в основном состоянии, поэтому при замещении водорода дейтерием реакция замедляется.
В пользу предположения, что главной причиной изотопных эффектов β-дейтерия является гиперконъюгация, свидетельствует тот факт, что максимальный эффект наблюдается, когда дейтерий находится в аятц-положении к уходящей группе (так как все атомы в резонансной системе должны быть копла-нарны, планарность системы О—С—С—X должна значительно увеличивать гиперконъюгацию), а также тот факт, что вторичные изотопные эффекты могут передаваться через ненасыщенные системы. Имеются данные о том, что по крайней мере некоторые изотопные эффекты β-дейтерия имеют стерическое происхождение (так, группа СD3 отличается меньшими стерическими требованиями, чем группа СН3); предлагалось также объяснение, основанное на учете эффектов поля (группа СD3 обладает, по-видимому, лучшими электронедонорными свойствами, чем группа СН3); тем не менее в большинстве случаев наиболее вероятной причиной этих эффектов следит считать гиперконъюгацию. Трудности объяснения вторичных изотопных эффектов отчасти связаны с их небольшой величиной, не превышающей обычно 1,5. Какова бы ни была причина вторичных изотопных эффектов β-дейтерия, их величина хорошо коррелирует с карбокатионным характером переходного состояния, и они служат полезным инструментом для исследования механизмов реакции.
Другого типа вторичные изотопные эффекты возникают в результате замещения водорода дейтерием у атома углерода, соединенного с уходящей группой. Эти вторичные изотопные эффекты α-дейтерия имеют величину от 0,87 до 1,26. Они также коррелируют с карбокатионным характером переходного состояния. В реакциях нуклеофильного замещения, где карбо-катионный интермедиат не образуется, изотопный эффект α-дейтерия близок к единице. В тех реакциях, в которых действительно промежуточно образуются карбокатпоны, наблюдается более высокий эффект, зависящий от природы уходящей группы. Природу изотопного эффекта α-дейтерия принято объяснять тем, что замещение водорода дейтерием оказывает более или менее сильное влияние па деформационные колебания связи С—Н в переходном, а не в основном состоянии, и в зависимости от природы переходного состояния скорость реакции может или увеличиваться, или уменьшаться.
Еще одним видом изотопных эффектов является изотопный эффект растворителя. Очень часто скорость реакции меняется при переходе от обычного растворителя к дейтерированному, например при замене Н2O на D2O или КОН на KОD. Возникающие изменения могут быть связаны с одним из трех перечисленных ниже факторов или с действием всех трех факторов.
1. Растворитель может одновременно быть реагентом, Если связь О—Н в молекуле растворителя разрывается в лимитирующей стадии, возникает первичный изотопный эффект. Если в реакции участвуют молекулы D2О или DзО+, может также наблюдаться вторичный изотопный эффект дейтерия, при котором связь D—О не разрывается,
2. За счет быстрого обмена водорода молекулы субстрата могут стать мечеными и разрываться в лимитирующей стадии.
3. Характер взаимодействий растворенное вещество—растворитель и степень этого взаимодействия могут быть различны в дейтерированном и в недейтерированиом растворителе; это может вызвать изменения в энергии переходного состояния, а, следовательно, и в энергии активации реакции. Тогда возникают вторичные изотопные эффекты. Для объяснения подобных эффектов предложены две физические модели. Очевидно, что во многих случаях одновременно действуют по крайней мере два фактора, первый и третий, а часто и все три. Предпринимались попытки разделить эти факторы.
Правило 1 Тетраэдрические системы.
а) 3—7-экзо-тет: выгодны
б) 5—6-эндо-тет: невыгодны
Правило 2. Тригональные системы.
а) 3—7-экзо-триг: выгодны
б) 3—5-эндо-триг: невыгодны
в) 6—7-эндо-триг: выгодны
Правило 3. Диагональные системы.
а) 3—4-экзо-диг: невыгодны
б) 5—7 экзо диг: выгодны
в) 3—7-эндо-диг: выгодны
«Невыгодный» не означает, что процесс не может быть осуществлен, просто он идет с большим трудом, чем «выгодный» процесс. Эти правила эмпирические и основаны на рассмотрении стереохимии, Выгодные пути замыкания цикла — это те, при которых длина и природа связывающего звена таковы, что конечные атомы могут достигнуть подходящей для реакции геометрии. При невыгодных путях требуется некоторое нарушение обычных валентных углов и межатомных расстояний. Многие известные примеры реакций замыкания цикла хорошо согласуются с правилами Болдуина.
Постулат Хэммонда
Поскольку переходные состояния имеют практически нулевое время жизни, их невозможно наблюдать непосредственно и об их геометрии можно только делать заключения на основании косвенных данных. Часто такие заключения бывают вполне основательны, Например, в реакции типа SN 2 между СН3I и I− (реакция, при которой продукт идентичен исходному соединению) переходное состояние должно быть совершенно симметричным. Однако во многих случаях невозможно прийти к таким легким выводам, и тогда на помощь приходит постулат Хэммонда, который гласит: геометрия переходного состояния похожа на геометрию тех веществ, к которым оно ближе по свободной энергии, и это относится к каждой стадии реакции. Так, в случае экзотермической реакции, переходное состояние больше похоже на реагенты, чем на продукты, хотя здесь разница не слишком велика, так как величина ΔG≠ с обеих сторон значительна. Этот постулат очень полезен при рассмотрении реакций, в ходе которых образуются интермедиаты. В реакции а, первое переходное состояние по энергии намного ближе к интермедиату, чем к реагентам, поэтому можно предполагать, что и геометрия его больше похожа на геометрию интермедиата, а не на геометрию реагентов, Точно так же второе переходное состояние по величине свободной энергии намного ближе к интермедиату, чем к продуктам, и потому по геометрии больше похоже на интермедиат, а не на продукты. О структуре пнтермедиатов обычно известно больше, чем о структуре переходных состояний, поэтому сведения об интермеднатах часто используются для того, чтобы сделать заключения о переходных состояниях
Дата добавления: 2020-02-05; просмотров: 239;
Поиск по сайту
Узнать еще
- F90 Гиперкинетические расстройства
- Аргументы должны быть достаточными для доказательства (быть соразмерными тезису).
- Аудиторские доказательства
- Аудиторские доказательства, методы их сбора.
- Вещественные доказательства.
- Виды доказательства
- Глава 47. Объекты биологического происхождения как вещественные доказательства
- Голосовые и кинетические средства воздействия

Публикации по технике и механике
Публикации по биологии

Публикации по информатике

Публикации по строительству
Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по истории