Полуэмпирическая квантовая химия
Принципиально иное направление, сыгравшее огромную роль в современном развитии химии, состоит в отказе от вычисления одноэлектронных и двуэлектронных интегралов, фигурирующих в методе ХФ. Вместо точного оператора Фока используется приближенный, элементы которого получают из эмпирических данных. Соответствующие параметры подбирают для каждого атома (иногда - с учетом конкретного окружения) и для пар атомов: таким образом, они являются либо фиксированными числами, либо зависят от расстояния между атомами При этом часто (но не обязательно - см. ниже) предполагается, что многоэлектронная волновая функция является однодетерминантной, а базисные функции À i – симметричными ортогональными комбинациями ОСТ {c j }. Такие комбинации получаются их исходных ОСТ с помощью преобразования À = c S-1/2 , где S– матрица интегралов перекрывания (эта процедура называется ортогоназизацией по Левдину). Расчет МО проводится обычным итерационным путем.
Полуэмпирические методы обычно на много порядков величины быстрее, чем неэмпирические, применимы к большим (часто к очень большим, например - биологическим) системам и для некоторых классов соединений они дают более точные результаты. Следует, однако, понимать, что это достигается за счет специально подобранных параметров, справедливых лишь в пределах узкого класса соединений. При переносе на другие классы, те же методы могут дать абсолютно неверные результаты. Кроме того, параметры расчета часто подбираются таким образом, чтобы воспроизводить те или иные молекулярные свойства, поэтому придавать физический смысл отдельным параметрам не следует. Основные приближения, используемые в полуэмпирических методах, следующие.
1. Рассматриваются только валентные электроны: считают, что электроны атомных остовов лишь экранируют ядра, поэтому эти электроны включают в функции, описывающие энергию отталкивания остов-остов (в которое включается ядерное отталкивание). Поляризацией остовов пренебрегают.
2. В МО учитывают только АО с главным квантовым числом, соответствующим высшим заселенным орбиталям изолированных атомов (минимальный базис), причем считают, что базисные функции образуют набор ортогональных АО.
3. Для двухэлектронных интегралов вводят приближение Нулевого Дифференциального Перекрывания (НДП)
c m ( r )c n ( r)dr =0. m ¹ n (2.44)
Считают, что из-за экспоненциального убывания АО двухэлектронными кулоновскими и обменными интегралами, содержащими произведения различных атомных орбиталей, зависящих от одного аргумента, можно пренебречь:
(m n ô l s ) = (m n ô l s )d m n d l s = (m m ô l l ) (2.45)
Это приближение позволяет резко уменьшить число двухэлектронных интегралов, которые необходимо вычислять, поэтому оно в том или ином виде используется во всех полуэмпирических методах.
В приближении НДП, принимаемом для всех пар АО, уравнения Рутана
имеют вид:
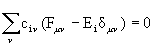 (2.46)
(2.46)
Элементы матрицы Фока записываются как
 ,
,
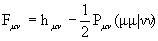 ,
, 
4. Результат расчета не должен зависеть от выбора декартовой системы коорди-нат, в которой определяются ориентации p-, d- и т.д. АО. НДП нарушает это требование. Рассмотрим двухцентровый кулоновский интеграл (px pe ï s2), где px и pe - АО одного и того же атома. В приближении НДП этот интеграл должен быть равен нулю. Повернем теперь декартову систему координат вокруг оси z на угол q : тогда
p¢ x = px cosq + py sinq (2.48)
p¢ y = px sinq - py cosq .
Интеграл (p¢ x p¢ e ï s2) в повернутой системе координат равен
(p¢ x p¢ e ï s2) = - (p 2x ï s2) cosq sinq + (p 2y ï s2) cosq sinq + (px pe ï s2)( cos2q - sin2q )=
= [(p 2y ï s2) - (p 2x ï s2) ] cosq sinq (2.49)
(учтено, что s-АО при повороте не меняется и что px и pe АО ортогональны).
Последнее равенство не равно нулю при произвольных значениях угла q - только при q , равном 00 или 900. Это означает, что рассматриваемый интеграл кулоновского отталкивания не инвариантен относительно поворота системы координат. Такое нарушение вращательной инвариантности имеет место каждый раз, когда двухэлектронные интегралы включают перекрывание двух АО одного и того же атома. Поэтому в таких случаях вводится еще одно приближение: считают, что двухэлектронные интегралы (m m ô l l ) зависят только от природы атомов, на которых центрированы орбитали c m и c l , и не зависят от конкретного вида орбиталей. Это соответствует сферическому усреднению распределения валентных электронов на АО различных атомов молекулы при расчете взаимодействия и обеспечивает инвариантность решения относительно поворота систем координат. Для усредненного интеграла (m m ô l l ) используется обозначение g AB , где А и В обозначают атомы, на которых центрированы АО интегралы m и l ; вычисляется он с s-АО соответствующих атомов
g AB = (s2Aô s2B) (2.50)
Что касается одноэлектронных интегралов, то для них также вводятся различные приближения, которые мы рассмотрим ниже применительно к конкретным методам. Остановимся здесь лишь на стандартных методах, получившим наибольшее распространение.
Дата добавления: 2022-04-12; просмотров: 101;
Поиск по сайту

Публикации по технике и механике
Публикации по биологии

Публикации по информатике

Публикации по строительству
Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по истории