Строение электронной оболочки ароматических систем.
Тенденция бензола и его производных к сохранению структуры сопряженного кольца в различных превращениях означает пвышенную термодинамическую и кинетическую. устойчивость этого структурного фрагмента. Стабилизация (понижение электронной энергии) молекулы или иона, обладающих циклической структурой, достигается при полном заполнении электронами всех связывающих молекулярных  орбиталей и вакантности несвязывающих и антисвязывающих орбиталей. Выполнение этих условий достигается, когда общее число
орбиталей и вакантности несвязывающих и антисвязывающих орбиталей. Выполнение этих условий достигается, когда общее число  электронов в циклич. полиене равно (4л + 2), где п = = 0,1,2... (правило Хюккеля).
электронов в циклич. полиене равно (4л + 2), где п = = 0,1,2... (правило Хюккеля).
Это правило объясняет устойчивость бензола (формула I) и циклопентадиенильного аниона (II; п = 1). Оно позволило правильно предсказать устойчивость циклопропенильного (III; п = 0) и циклогептатриенильного (IV; п = 1) катионов. Ввиду подобия  электронных оболочек соединений II-IV и бензола они, как и высшие циклич. полиены - [10], [14], [18]аннулены (V-VII), рассматриваются как ароматические системы.
электронных оболочек соединений II-IV и бензола они, как и высшие циклич. полиены - [10], [14], [18]аннулены (V-VII), рассматриваются как ароматические системы.

Правило Хюккеля можно экстраполировать на ряд сопряженных гетероциклических ссоединений. - производные пиридина (VIII) и катиона пирилия (IX), изоэлектронные бензолу, пятичленные гетероциклы типа X (пиррол, фуран, тиофен), изоэлектронные циклопентадиенильному аниону. Эти соединения также относят к ароматическим. системам.

Для производных соединений II-Х и др. более сложных структур, получаемых изоэлектронным замещением метиновых групп в полиенах I-VII, также характерны высокая термодинамическая устойчивость и общая склонность к реакциям замещения в ядре.
Циклически сопряженные полиены, имеющие в цикле 4n  электронов (n=1,2...), неустойчивы и легко вступают в реакции присоединения, т. к. обладают незамкнутой электронной оболочкой с частично заполненными несвязывающими орбиталями. Такие соединения, наиб. типичным примером которых служит циклобутадиен (XI), относят к антиароматическим системам.
электронов (n=1,2...), неустойчивы и легко вступают в реакции присоединения, т. к. обладают незамкнутой электронной оболочкой с частично заполненными несвязывающими орбиталями. Такие соединения, наиб. типичным примером которых служит циклобутадиен (XI), относят к антиароматическим системам.
Правила, учитывающие число  электронов в цикле, полезны для характеристики свойств моноциклических структур структур, однако неприложимы к полициклам. При оценке ароматичности последних необходимо учитывать, как соответствуют этим правилам
электронов в цикле, полезны для характеристики свойств моноциклических структур структур, однако неприложимы к полициклам. При оценке ароматичности последних необходимо учитывать, как соответствуют этим правилам  электронные оболочки каждого отдельного цикла молекулы. С осторожностью следует пользоваться ими и в случае многозаряженных циклич. ионов. Так, электронные оболочки дикатиона и дианиона циклобутадиена отвечают требованиям правила Хюккеля. Однако эти структуры нельзя относить к ароматическим, т. к. дикатион (п = 0) устойчив не в плоской форме, обеспечивающей циклич. сопряжение, а в согнутой по диагонали; дианион (n=1) вообще неустойчив.
электронные оболочки каждого отдельного цикла молекулы. С осторожностью следует пользоваться ими и в случае многозаряженных циклич. ионов. Так, электронные оболочки дикатиона и дианиона циклобутадиена отвечают требованиям правила Хюккеля. Однако эти структуры нельзя относить к ароматическим, т. к. дикатион (п = 0) устойчив не в плоской форме, обеспечивающей циклич. сопряжение, а в согнутой по диагонали; дианион (n=1) вообще неустойчив.
Энергетические критерии ароматичности. Энергия резонанса.Для определения количеств. меры ароматичности, характеризующей повыш. термодинамич. устойчивость ароматич. соед., было сформулировано понятие энергии резонанса (ЭР), или энергии делокализации.
Теплота гидрирования молекулы бензола, формально содержащей три двойные связи, на 151 кДж/моль больше, чем теплота гидрирования трех молекул этилена. Эту величину, связываемую с ЭР, можно рассматривать как энергию, дополнительно затрачиваемую на разрушение циклич. системы сопряженных двойных связей бензольного кольца, стабилизирующей эту структуру. Т. обр., ЭР характеризует вклад циклич. сопряжения в теплоту образования (полную энергию, теплоту атомизации) соединения.
Предложен ряд способов теоретич. оценок ЭР. Они различаются гл. обр. выбором структуры сравнения (т.е. структуры, в к-рой нарушено циклич. сопряжение) с циклич. формой. Обычный подход к вычислению ЭР состоит в сопоставлении  электронных энергий
электронных энергий  циклич. структуры и суммы энергий всех изолированных кратных связей, содержащихся в ней. Однако рассчитываемые т. обр. ЭР, независимо от используемого квантовохим. метода, имеют тенденцию к возрастанию с увеличением размеров
циклич. структуры и суммы энергий всех изолированных кратных связей, содержащихся в ней. Однако рассчитываемые т. обр. ЭР, независимо от используемого квантовохим. метода, имеют тенденцию к возрастанию с увеличением размеров  системы. Это нередко противоречит эксперим. данным о св-вах ароматич. системы. Так, ароматичность в ряду полиаценовбензол (I), нафталин (XII), антрацен (XIII), тетрацен (XIV) понижается (напр., возрастает склонность к присоединению, увеличивается альтернирование длин связей), а ЭР (приведены в единицах
системы. Это нередко противоречит эксперим. данным о св-вах ароматич. системы. Так, ароматичность в ряду полиаценовбензол (I), нафталин (XII), антрацен (XIII), тетрацен (XIV) понижается (напр., возрастает склонность к присоединению, увеличивается альтернирование длин связей), а ЭР (приведены в единицах  = 75 кДж/моль) растут:
= 75 кДж/моль) растут:

Этого недостатка лишены величины ЭР, рассчитываемые путем сравнения 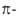 электронных энергий циклич. структуры и аналогичного ациклич. сопряженного полнена (М. Дьюар, 1969). Рассчитанные т. обр. величины принято называть ЭР Дьюара (ЭРД). Напр., ЭРД бензола (1,013) вычисляется при сопоставлении его
электронных энергий циклич. структуры и аналогичного ациклич. сопряженного полнена (М. Дьюар, 1969). Рассчитанные т. обр. величины принято называть ЭР Дьюара (ЭРД). Напр., ЭРД бензола (1,013) вычисляется при сопоставлении его 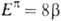 с
с 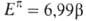 1,3,5-гексатриена, а ЭРД циклобутадиена
1,3,5-гексатриена, а ЭРД циклобутадиена  -сопоставлением его
-сопоставлением его  = =
= = 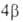 с
с  1,3-бутадиена.
1,3-бутадиена.
Соединения с положит. значениями ЭРД относят к ароматическим, с отрицательными-к антиароматическим, а со значениями ЭРД, близкими к нулю, - к неароматическим. Хотя значения ЭРД варьируют в зависимости от приближений квантовохим. метода расчета, относит. порядок их практически не зависит от выбора метода. Ниже приведены ЭРД в расчете на один 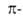 электрон (ЭРД/е; в единицах
электрон (ЭРД/е; в единицах 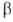 ), вычисленные по модифициров. методу молекулярных орбиталей Хюккеля:
), вычисленные по модифициров. методу молекулярных орбиталей Хюккеля:

Наиб. ЭРД/е, то есть наиб. ароматичностью, обладает бензол. Понижение ЭРД/е отражает понижение ароматич. св-в. Приведенные данные хорошо согласуются со сложившимися представлениями о проявлениях ароматичности.
Магнитные критерии ароматичности.Циклич. сопряжение  электронов приводит к возникновению в молекуле кольцевого тока, к-рый вызывает экзальтацию диамагн. восприимчивости. Поскольку величины кольцевого тока и экзальтации отражают эффективность циклич. сопряжения, они м. б. использованы как количеств. мера ароматичности.
электронов приводит к возникновению в молекуле кольцевого тока, к-рый вызывает экзальтацию диамагн. восприимчивости. Поскольку величины кольцевого тока и экзальтации отражают эффективность циклич. сопряжения, они м. б. использованы как количеств. мера ароматичности.
К ароматическим относятся соед., в молекулах к-рых поддерживаются наведенные диамагнитные  электронные кольцевые токи (диатропные системы). В случае [4/1 + 2]аннуленов (n = 0,1,2...) существует прямая пропорциональность между силой кольцевого тока и величиной ЭРД. Однако для неальтернантных углеводородов (напр., азулена) и гетероциклич. соед. эта зависимость усложняется. В ряде случаев система м.б. одновременно и диатропной и антиароматической, напр. бицикло[6,2,0]декапентаен. Наличие индуциров. кольцевого тока в циклич. сопряженных системах характерно проявляется в спектрах протонного магн. резонанса (ПМР), т.к. ток создает анизотропное магн. поле, заметно влияющее на хим. сдвиги протонов, связанных с атомами кольца. Сигналы протонов, расположенных во внутр. части ароматич. кольца, смещаются в сторону сильного поля, а сигналы протонов, расположенных на периферии кольца, - в сторону слабого поля. Так, внутр. протоны [14]аннулена (ф-ла VI) и [18]аннулена (VII) проявляются при — 60°С в спектре ПМР соотв. при 0,0 и —2,99м. д., а внешние-при 7,6 и 9,28 м. д. Для антиароматич. систем [4n]аннуленов, наоборот, характерны парамагн. кольцевые токи, приводящие к сдвигу внеш. протонов в сильное поле (паратропные системы). Так, хим. сдвиг внеш. протонов [16]аннулена равен всего 4,8 м.д.
электронные кольцевые токи (диатропные системы). В случае [4/1 + 2]аннуленов (n = 0,1,2...) существует прямая пропорциональность между силой кольцевого тока и величиной ЭРД. Однако для неальтернантных углеводородов (напр., азулена) и гетероциклич. соед. эта зависимость усложняется. В ряде случаев система м.б. одновременно и диатропной и антиароматической, напр. бицикло[6,2,0]декапентаен. Наличие индуциров. кольцевого тока в циклич. сопряженных системах характерно проявляется в спектрах протонного магн. резонанса (ПМР), т.к. ток создает анизотропное магн. поле, заметно влияющее на хим. сдвиги протонов, связанных с атомами кольца. Сигналы протонов, расположенных во внутр. части ароматич. кольца, смещаются в сторону сильного поля, а сигналы протонов, расположенных на периферии кольца, - в сторону слабого поля. Так, внутр. протоны [14]аннулена (ф-ла VI) и [18]аннулена (VII) проявляются при — 60°С в спектре ПМР соотв. при 0,0 и —2,99м. д., а внешние-при 7,6 и 9,28 м. д. Для антиароматич. систем [4n]аннуленов, наоборот, характерны парамагн. кольцевые токи, приводящие к сдвигу внеш. протонов в сильное поле (паратропные системы). Так, хим. сдвиг внеш. протонов [16]аннулена равен всего 4,8 м.д.
Структурные критерии ароматичности.Важнейшие структурные характеристики молекулы бензола - ее планарность и полная выравненность связей. Молекулу можно рассматривать как ароматическую, если длины углерод-углеродных связей в ней лежат в пределах 0,136-0,143 нм, т.е. близко к 0,1397 нм для молекулы бензола (I). Для нециклических сопряженных полиеновых структур длины связей С—С составляют 0,144-0,148 нм, а связей С=С-0,134-0,135 нм. Еще большее альтернирование длин связей характерно для антиароматич. структур. Это подтверждается данными строгих неэмперических расчетов геометрических параметров циклобутадиена и эксперим. данными для его производных.
Предложены разл. выражения для количеств. характеристики ароматичности по степени альтернирования длин связей, напр. для углеводородов вводится индекс ароматичности (НОМАd):

где а = 98,89, Хr- длина r-ной связи (в А), n-число связей. Для бензола HOMAd максимален и равен 1, для циклобутадиена минимален (0,863). Азулен с НОМАd = 0,921 занимает промежут. положение, характерное для неароматич. систем.
Развитие концепции ароматичности.Главная характеристика - энергетич. стабилизация структуры при молекулярной геометрии, создающей оптимальные условия для соответствующих электронных взаимодействий. Установление аналогичных связей между пространственным и электронным строением молекул др. структурных типов привело к расширению понятия ароматичности. Повыш. устойчивость гомосопряженных систем (относительно др. изомерных форм), в к-рых число 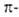 электронов равно (4n + 2), привела к выработке понятия гомоароматичности. Представителями гомоароматич. систем служат, напр., 1,3,5-циклогептатриен (гомобензол; XV) и трис-гомоциклопропенильный катион (XVI).
электронов равно (4n + 2), привела к выработке понятия гомоароматичности. Представителями гомоароматич. систем служат, напр., 1,3,5-циклогептатриен (гомобензол; XV) и трис-гомоциклопропенильный катион (XVI).

К спироароматич. системам принадлежат ненасыщ. спирановые структуры, в к-рых перекрывание двух ортогональных систем  орбиталей приводит к стабилизирующему эффекту. Последний достигается, когда число
орбиталей приводит к стабилизирующему эффекту. Последний достигается, когда число  электронов в обоих циклах равно (4п + 2), напр. в [4,2]спирарене (XVII). Понятие ароматичности привлекается даже для характеристики энергетически устойчивых нециклич. структур [напр., дианиона триметилена и его изоэлектронного аналога гуанидина (ХУШ)]-Y-ароматичность; для насыщ. циклов [напр., циклоалканов с нечетным числом метиленовых звеньев, как циклопропан (XIX)] - сигма-ароматичность; трехмерных каркасных структур - трехмерная ароматичность.
электронов в обоих циклах равно (4п + 2), напр. в [4,2]спирарене (XVII). Понятие ароматичности привлекается даже для характеристики энергетически устойчивых нециклич. структур [напр., дианиона триметилена и его изоэлектронного аналога гуанидина (ХУШ)]-Y-ароматичность; для насыщ. циклов [напр., циклоалканов с нечетным числом метиленовых звеньев, как циклопропан (XIX)] - сигма-ароматичность; трехмерных каркасных структур - трехмерная ароматичность.

Примеры структур с трехмерной ароматичностью-углеводородные катионы (СН)5+ (ф-ла XX), (СН)62+ (XXIX производные к-рых известны, нидо- и клозокарбораны (XXII и ХХIII),  комплексы типа железа карбонилов и др. (см. Карбонилы металлов), сэндвичевые структуры типа ферроцена, металлоорг. кластеры-производные переходных металлов. Во всех этих структурах реализуется замкнутая оболочка валентных электронов, заполняющих только связывающие молекулярные орбитали. Для разл. типов каркасных структур, напр. пирамидальных, сэндвичевых, бипирамидальных, разработаны специфич. правила электронного счета, определяющие их устойчивость, т.е. ароматичность.
комплексы типа железа карбонилов и др. (см. Карбонилы металлов), сэндвичевые структуры типа ферроцена, металлоорг. кластеры-производные переходных металлов. Во всех этих структурах реализуется замкнутая оболочка валентных электронов, заполняющих только связывающие молекулярные орбитали. Для разл. типов каркасных структур, напр. пирамидальных, сэндвичевых, бипирамидальных, разработаны специфич. правила электронного счета, определяющие их устойчивость, т.е. ароматичность.

Понятие ароматичности успешно привлекается для описания энергетич. характеристик переходных состояний термич. перициклич. р-ций. Такие р-ции осуществляются через переходные состояния, к-рые в зависимости от конформации цикла содержат 4n+2 (хюккелевские системы) или 4n (мебиусовские системы) 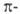 электронов.
электронов.
Вопрос 7. Электронодонорные и электроноакцепторные заместители в ароматическом кольце.
Важнейшим фактором, определяющим химические свойства молекулы, является распределение в ней электронной плотности. Характер распределения зависит от взаимного влияния атомов.В молекулах, имеющих только s -связи, взаимное влияние атомов осуществляется через индуктивный эффект. В молекулах, представляющих собой сопряженные системы, проявляется действие мезомерного эффекта.Влияние заместителей, передающееся по сопряженной системе p -связей, называется мезомерным (М) эффектом.В молекуле бензола p -электронное облако распределено равномерно по всем атомам углерода за счет сопряжения. Если же в бензольное кольцо ввести какой-нибудь заместитель, это равномерное распределение нарушается и происходит перераспределение электронной плотности в кольце. Место вступления второго заместителя в бензольное кольцо определяется природой уже имеющегося заместителя.Заместители подразделяют на две группы в зависимости от проявляемого ими эффекта (мезомерного или индуктивного): электронодонорные и электроноакцепторные.Электронодонорные заместители проявляют +М- и +I-эффект и повышают электронную плотность в сопряженной системе. К ним относятся гидроксильная группа —ОН и аминогруппа —NН2. Неподеленная пара электронов в этих группах вступает в общее сопряжение с p -электронной системой бензольного кольца и увеличивает длину сопряженной системы. В результате электронная плотность сосредоточивается в орто- и пара-положениях:
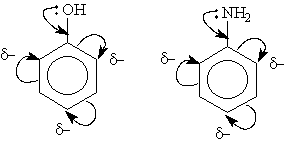
Алкильные группы не могут участвовать в общем сопряжении, но они проявляют +I-эффект, под действием которого происходит аналогичное перераспределение p -электронной плотности.
Электроноакцепторные заместители проявляют -М-эффект и снижают электронную плотность в сопряженной системе. К ним относятся нитрогрупла —NO2, сульфогруппа —SO3Н, альдегидная —СНО и карбоксильная —СООН группы. Эти заместители образуют с бензольным кольцом общую сопряженную систему, но общее электронное облако смещается в сторону этих групп. Таким образом, общая электронная плотность в кольце уменьшается, причем меньше всего она уменьшается в мета-положениях:

Полностью галогенизированные алкильные радикалы (например, —ССl3) проявляют -I-эффект и также способствуют понижению электронной плотности кольца.
Закономерности преимущественного направления замещения в бензольном кольце называют правилами ориентации.
Заместители, обладающие +I-эффектом или +М-эффектом, способствуют электрофильному замещению в орто- и пара-положениях бензольного кольца и называются заместителями (ориентантами) первого рода:

Заместители, обладающие -I-эффектом или -М-эффектом, направляют электрофильное замещение в мета-положения бензольного кольца и называются заместителями (ориентантами) второго рода:
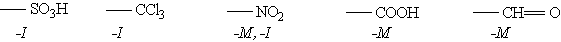
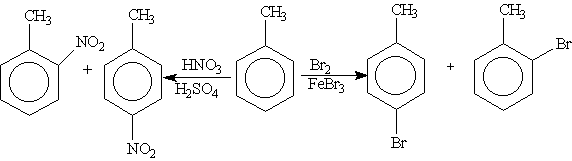
Так, толуол, содержащий заместитель первого рода, нитруется и бромируется в пара- и орто-положения:
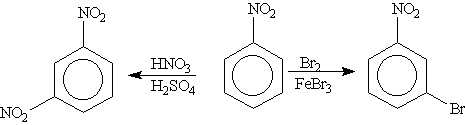
Нитробензол, содержащий заместитель второго рода, нитруется и бромируется в мета-положение:
Помимо ориентирующего действия заместители оказывают влияние и на реакционную способность бензольного кольца: ориентанты 1-го рода (кроме галогенов) облегчают вступление второго заместителя; ориентанты 2-го рода (и галогены) затрудняют его.
Вопрос 8. Кислтность и основность органических молекул.
Существуют два общепринятых способа определения понятий кислота и основание – определения Бренстеда и Льюиса.
Бренстедовская кислотность и основность органических соединений
По Бренстеду кислоты – это соединения, способные к отдаче протона, а основания – соединения, способные принимать протон.
Бренстед Йоханнес Николаус
 (22.II.1879–17.XII.1947) (22.II.1879–17.XII.1947)
| Датский физикохимик, член Датского королевского общества наук. Основные работы посвящены химической кинетике, катализу и термодинамике растворов. Сформулировал (1929) основные положения «общей», или «расширенной», теории кислот и оснований, согласно которой: а) кислота является донором, а основание – акцептором протона; б) кислоты и основания существуют только как сопряженные пары; в) протон не существует в растворе в свободном виде, в воде он образует ион |
Любое вещество может рассматриваться как потенциальная кислота, с отщеплением протона превращающаяся в сопряженное данной кислоте основание.
Точно так же любое вещество может рассматриваться и как основание, после присоединения протона превращающееся в сопряженную данному основанию кислоту:
АН + В ↔ А– + НВ+
кислота основание основание кислота
Кислота АН и основание А–, так же как и НВ+ и В, являются сопряженными кислотно-основными парами.

Для диссоциации слабых электролитов в разбавленных водных растворах мерой кислотности является положение равновесия в реакции:
АН ↔H+ + A–
Константа равновесия (диссоциации) для этой реакции:

Эта величина, как правило, много меньше единицы (даже для СH3СОOH Ка = 1,76٠10-5 = 10-4,7). Для большего удобства вводят понятие величины pKa:
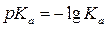
Чем меньше рКа, тем больше кислотность соединения по Бренстеду. Например: e метана рКа= 40, у метанола 16, у воды 14, у фенола 9,8, у уксусной кислоты 4,7, у соляной кислоты меньше 1.
Таблица 1
Значение рКа некоторых органических соединений
| Кислота | рКа |
| Хлорная Азотная Соляная Серная Бензолсульфокислота Тринитрометан | <0 Сильные кислоты |
| Пикриновая Трихлоруксусная Щавелевая Сернистая Фосфорная Хлоруксусная Азотистая Бензойная Уксусная Пиридиний-катион Лимонная Сероводородная Борная Аммоний-ион Фенол Нитрометан Этилмеркаптан Вода Этиловый спирт Ацетон Анилин Аммиак Толуол Метан | 0,4 0,7 1,2 1,8 2,1 2,9 3,3 4,2 4,7 5,2 5,4 7,2 9,2 9,2 9,9 11,0 12,0 14,0 18,0 20,0 27,0 30,0 35,0 40,0 |
Основность соединений оценивают по величине рКа сопряженных с ними кислот. Чем больше рКа, сопряженной кислоты, тем больше основность.
Например: Для метиламина СH3NH2, оценивают рКа сопряженной с ним кислоты:
СH3NH3 ↔ СH3NH2 + H+
Для оценки основности иногда используют и величину рКb:
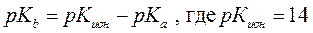
Например: для уксусной кислоты рКа = 4,7, значит рКb = 14 – 4,4 = 9,3 (основность сопряженного основания, т.е. ацетат-иона).
Если изменяется атом, по связи которого с атомом водорода происходит ионизация, то чем больше прочность этой связи, тем меньше должна быть константа диссоциации. Такое соответствие действительно имеет место при сопоставлении кислот НХ с атомами X, относящимися к элементам одной и той же группы.
Таблица 2
рКа некоторых соединений
| Связь | O–H | S–H | N–H | Р–Н |
| Есв,кДж/моль | 459,8 | 365,7 | 349,9 | 263,3 |
| рКа | 16(H2O) | 7 (H2S) | 35 (NH3) | 27(РH3) |
При сравнении же рКа, соединений НХ, имеющих атомы X, которые относятся к элементам разных групп, но одного и того же периода, видно, что такое рассмотрение является упрощенным, так как природа атома X имеет существенное значение. Элементы одного периода имеют тем большее сродство к электрону, чем больше аффективный заряд ядра, и этот фактор является определяющим. В результате H2O и H2S – более сильные кислоты, чем NH3 и РH3соответственно, хотя энергии разрыва связей в первых соединениях больше, чем в последних.
Для определения влияния заместителей на константы диссоциации органических кислот берут серию кислот, в которых остается неизменным скелет молекулы и меняется лишь заместитель. Обычно оценивается влияние заместителей на изменение Ка замещенных кислот по сравнению с Ка незамещенной кислоты (водные растворы, 25°С).
При рассмотрении влияния заместителей можно вначале, не учитывая участия растворителя, проанализировать влияние заместителей на Ка соответственно замещенной кислоты в газовой фазе и, используя представления об электронных эффектах заместителей и о механизме передачи этих эффектов к реакционному центру, представить себе внутреннюю для данной молекулы причину изменения Ка, при смене заместителей.
Рассмотрим влияние заместителей на изменение Ка, в серии замещенных уксусных кислот. Большие константы ионизации хлор- и бромуксусной кислот по сравнению с фторуксусной объясняются тем, что в газовой фазе (в отсутствие растворителя) стабилизация аниона осуществляется внутримолекулярно и, чем больше поляризуемость примыкающего к анионному центру остатка (Вr > Сl > F), тем стабильнее анион.
Сильное влияние на константы диссоциации может оказывать заместитель, если он способен к непосредственному взаимодействию с карбоксильной группой с образованием псевдоциклических соединений. Одним из таких хорошо изученных взаимодействий является образование водородной связи. Последняя может образовываться в зависимости от природы заместителя либо при участии кислородного атома карбоксильной группы, либо за счет водородного атома той же группы. Первое взаимодействие приводит к увеличению Ка, второе – к уменьшению Ка, по сравнению с соединением, не имеющим заместителя.
Например: Вследствие образования водородной связи Ка cалициловой кислоты много больше, чем Ка изомерных ей м- и п-оксибензойных кислот:
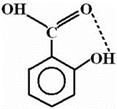
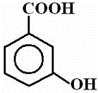

рКа= 2.98 рКа= 4.06 рКа= 4.48
Увеличение константы диссоциации обуславливается большей энергией образования водородной связи в анионе салициловой кислоты, чем в недиссоциировапной молекуле. Таким образом, водородная связь больше стабилизирует конечное состояние, чем исходное.
Если образование внутримолекулярной водородной связи происходит при участии водорода карбоксильной группы, как это имеет место в о-метоксибензойной кислоте (I), то, наоборот, стабилизируется исходное состояние и вследствие этого константа диссоциации уменьшается (рКа =4,02); при этом возможно, что наблюдаемое значение рКа, в основном обусловлено существованием в равновесии открытой формы (II):

(I) (II)
Если в молекуле имеется две карбоксильные группы, то кислота характеризуется двумя константами диссоциации К1 и К2. К1 т.е. константа диссоциации молекулы всегда больше, чем константа диссоциации образовавшегося иона.
Например:СООНрК1=1,23
|
СООНрК2=4,19
Кроме электронного влияния, передаваемого в основном по линии ковалентных связей, заместитель может оказывать влияние на константы ионизации органических кислот при введении объемистых заместителей, особенно на близком расстоянии от карбоксильной группы, так как заместители будут создавать препятствия для сольватации как кислоты, так и в большей степени соответствующего аниона. Это должно дестабилизировать конечное состояние больше, чем исходное. В результате это приводит к снижению константы диссоциации, что и наблюдается в алифатическом ряду при увеличении разветвления цепи.
Спирты диссоциированы в водных растворах значительно меньше, чем кислоты, что связано с меньшей делокализацией отрицательного заряда.
Фенолы диссоциированы на ионы в водном растворе в значительно меньшей степени, но влияние заместителей проявляется сильнее, особенно когда последние являются элекгроноакцепторами и характеризуются большим –М-эффектом.
Многие соединения с этих позиций являются амфотерными.
Кислотность и основность по Бренстеду является частным случаем более широкого представления о кислотах и основаниях, данного Льюисом.
В отличие от Бренстеда, положившего в основу характеристики кислотно-основных взаимодействий перенос протона от кислоты к основанию, Льюис предложил оценивать кислотные и основные свойства органических соединений по способности принимать или предоставлять электронную пару с последующим образованием ковалентной связи.
 Льюис Гильберт Ньютон
(23.X.1875–23.III.1946) Льюис Гильберт Ньютон
(23.X.1875–23.III.1946)
| Американский физикохимик. Основные работы посвящены химической термодинамике и теории строения вещества. Определил свободную энергию многих соединений. Ввел понятие термодинамической активности. Уточнил формулировку закона действующих масс. Развил (1916) теорию ковалентной химической связи. Его концепция обобщенной электронной пары оказалась очень плодотворной для органической химии. Предложил (1926) новую теорию кислот как акцепторов пары электронов и оснований как доноров пары электронов. Ввел (1929) термин «фотон». |
Атом, принимающий электронную пару, является акцептором, электронов и соединение, имеющее такой атом, функционирует как кислота.
Атом, предоставляющий неподеленную электронную пару, является донором электронов и соединение, имеющее такой атом, является основанием.
Более общая формулировка Льюиса позволяет рассматривать большое число органических реакций, в которых участвуют кислоты Льюиса – электрофильные реагенты или основания Льюиса – нуклеофильные реагенты с единой позиции кислотно-основных взаимодействий.
В результате взаимодействия кислоты и основания Льюиса на атоме-акцепторе электронов возникает отрицательный заряд, а на атоме-доноре электронов положительный заряд, и образуется либо соединение, содержащее координационную связь, либо комплексы, называемые, в соответствии с механизмом их образования, электронодонорно-акцепторными или просто донорно-акцепторными.
Кислотами Льюиса являются частицы, содержащие низкие по энергии вакантные орбитали (H+, BF3, АlСl3 катионы и галогениды переходных металлов Аg+,SnСl4), а также соединения, содержащие π-связи, сопряженные с заместителями с большим –М-эффектом.
Основаниями Льюиса являются соединения, содержащие атомы с неподеленными электронными парами или π-связи (в том числе ароматические и гетероциклические соединения), донорная способность которых увеличена заместителями, проявляющими +М-эффект.
Кислоты Льюиса (AlBr3, ZnСl2, ВF3) реагируют с основаниями Льюиса (Вr–, Сl–, R2О).
Льюисовские кислотность и основность органических соединений могут быть оценены лишь относительно – путем определения энергии взаимодействия различных соединений с одним и тем же стандартным соединением, являющимся соответственно основанием или кислотой Льюиса. Все остальные факторы (в частности, растворитель, температура) должны быть одинаковыми.
Ряд относительности кислот и оснований Льюиса не носит столь универсального характера, как для кислот и оснований Бренстеда. Относительная основность оснований Льюиса может существенно зависеть от того, какая кислота принята за стандарт.
Следует заметить, что кислотность и основность сильно зависят от растворителя. Обычно в растворителе, кислотность которого выше кислотности воды, сила кислот уменьшается, а сила оснований возрастает. В растворителе с более высокой по сравнению с водой основностью сила кислот возрастает, а сила оснований уменьшается.
Льюисовские кислоты и основания согласно Пирсону, могут быть подразделены на два типа: жесткие и мягкие.
Донорные атомы жестких оснований характеризуются высокой электроотрицательностыо, низкой поляризуемостью, они с трудом теряют свои электроны и поэтому трудно окисляются.
Донорные атомы мягких оснований имеют малую электроотрицательность, большую поляризуемость, легко окисляются.
На основании этих характеристик Пирсон предложил следующую классификацию оснований:
Таблица 3
Классификация оснований по Пирсону
| Жесткие | Пограничные | Мягкие |
| Н2O РO43– | С6Н5NН2 | R2S СN– |
| НО– СlO4– | С5Н5N | RCN СО |
| СН3СОО– Сl– | N3– | R2S– Н+ |
| RО– RОН | Вr– | I– R+ |
| R2O NН3 | SСN– С2Н4 | |
| RNН2 F– | R3Р С6Н6 | |
| N2H4 |
Жесткие основания легко образуют водородные связи.
Аналогично кислоты Льюиса могут быть подразделены на жесткие и мягкие.
Акцепторные атомы жестких кислот обладают большим положительным зарядом, имеют малый объем, низкую поляризуемость, высокую электроотрицательность.
Мягкие кислоты имеют акцепторный атом большего объема с малой электроотрицательностью, высокую поляризуемость.
Самой жесткой кислотой является протон, самые мягкие кислоты – катионы переходных металлов.
На основании этих характеристик Пирсон предложил следующую классификацию кислот:
Таблица 4
Классификация кислот по Пирсону
| Жесткие | Пограничные | Мягкие |
| H+ | Zn2+ | Сu2+ |
| Li+ | Sn2+ | Аg+ |
| Na+ | В(СН3)3 | Hg+ |
| К+ | R3C+ | Рd2+ |
| Mg2+ | SО2 | CH3Hg+ |
| Ca2+ | I2 | |
| Аl3+ | Вr2 | |
| ВF3 | IСN | |
| АlСl3 | Карбены | |
| Тринитробензол | ||
| R – С = О | Хиноны |
Жесткие кислоты более энергично и с образованием более прочных соединений реагируют с жесткими основаниями, а мягкие кислоты – с мягкими (принцип жестких и мягких кислот и оснований – ЖМКО). Это правило было установлено при измерении относительной устойчивости комплексных соединений. Образование более устойчивых комплексов при взаимодействии кислот и оснований одинакового характера было названо «симбиозом».
Относительная мягкость или жесткость кислот и оснований зависит от растворителя. Растворители, хорошо сольватирующие жесткие анионы, т.е. анионы малого размера, уменьшают их активность и делают менее жесткими.
Мягкие основания сольватируются теми же растворителями значительно меньше и вследствие этого различия в жесткости и мягкости растворителей такого типа становятся менее выраженными.
Изложенные представления широко используются в неорганической химии, а также при рассмотрении процессов сольватации. В последнее время теорию ЖМКО стали применять и для интерпретации данных по равновесным органическим процессам.
. Химические свойства органических кислот и оснований
(реакционная способность и качественные реакции)
Химические реакции, характерные для гидроксилсодержащих соединений. Гидроксилсодержащие соединения имеют в своем составе одну или несколько гидроксильных групп; к ним относятся одно- и многоатомные спирты, фенолы и др.
а)Окисление спиртов осуществляется в достаточно жестких условиях (t°, кислая cреда): первичные спирты окисляются до альдегидов, вторичные — до кетонов, третичные — не окисляются (в стандартных условиях)
2K2Cr2O7 + 2H2SO4 + H2O ® 2K2SO4 + 2H2CrO4 + H2Cr2O7

б) Качественная реакция на многоатомные спирты (глицерин) связана с тем, что в растворе многоатомного спирта диссоциирует, как правило, одна гидроксильная группа, а остальные недиссоциированные гидроксильные группы увеличивают стабильность аниона многоатомного спирта. В связи с этим атом водорода в -ОН-кислотном центре многоатомного спирта способен замещаться на атом меди при взаимодействии с гидроксидом меди (II), давая растворимый окрашенный комплекс.

в) Кислотность фенолов значительно выше, чем у спиртов, т. к. стабильность аниона значительно повышается за счет делокализации отрицательного заряда по системе сопряженных связей ароматического ядра.

Фенол, являясь более сильной кислотой, чем алифатические спирты (этанол, в частности), но слабее минеральных кислот, способен реагировать с гидроксидом натрия, образуя растворимый в воде фенолят натрия.
С6Н5ОН + NaOH ® C6H5ONa +H2O
Но при добавлении разбавленной Н2SO4 вновь выделяется фенол (образуется эмульсия).
С6Н5ОNa + Н2SO4 ® C6Н5ОН + NaHSO4
г) Качественная реакция на фенол — качественная реакция на гидроксил, связанный с ненасыщенным атомом углерода обусловлена тем, что водород в ОН-кислотном центре фенола способен замещаться на атом железа, так как фенолы — более сильные кислоты, чем спирты. Соответствующий феноксид — анион стабилизируется за счет р-p-p-p-сопряжения. При образовании фенолята железа d-орбитали иона Fe3+ вступают в сопряжение с системой сопряжения в феноксид-анионе, что приводит к удлинению системы сопряжения, в результате соединение становится окрашенным, т. к. поглощает свет в видимой области

Химические реакции аминопроизводных алифатического и ароматического рядов.
Алифатические радикалы, обладая положительным индуктивным эффектом +I, увеличивают электронную плотность на атоме азота и, соответственно, способность аминогруппы связывать протон (Н+), поэтому алифатические амины более сильные основания, чем аммиак NН3.

В ароматических аминах, в анилине, в частности, неподеленная электронная пара атома азота находится в сопряжении с p-электронной системой ароматического ядра, она менее доступна для образования связи с протоном, поэтому анилин — более слабое основание, чем аммиак и, тем более, чем алифатический амин — метиламин.

Вопрос 9. Понятие о С–Н, О–Н, N–Н, S–Н кислотах.
СН-КИСЛОТЫ, органические соединения в которых под действием оснований или. физических факторов (напр., ионизирующего излучения) может происходить гетеролиз связи С—Н с образованием протона и карбаниона. Известны также ОН-, SH-, NH-, SiH-кислоты и т.д., которые при отрыве протона образуют соответствующие анионы. СН-кислотами могут быть насыщенные и ненасыщенные углеводороды ациклического и циклического строения, напр. пропилен, ацетилен, толуол, трифенилметан, инден, циклопентадиен, ацетоуксусный и малоновый эфиры, HCN, а также заряженные частицы, напр. аренониевые ионы (см. Ареноний-кат ионы). Количеств, характеристикой СН-кислотности служит константа равновесия К реакции:
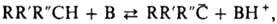
где В-основание (растворитель). Обычно используют значение рКа=-lgK. Аналогично характеризуют кислотность ОН-кислот и др. Для определения рКаприменяют индикаторный, полярографич. и электрохим. методы, метод переметаллирования, ионный циклотронный резонанс и др. Наличие единого параметра позволяет сопоставлять термодинамич. стабильность образующихся анионов в выбранном р-рителе. Существуют шкалы кислотности в воде, ДМСО, N-метилп
Дата добавления: 2016-11-04; просмотров: 1539;
Поиск по сайту
Узнать еще
- D-технология построения чертежа. Типовые объемные тела: призма, цилиндр, конус, сфера, тор, клин. Построение тел выдавливанием и вращением. Разрезы, сечения.
- I. Синусы твердой оболочки головного мозга.
- II. Митохондрии (строение и функции)
- II. Построение продольного профиля по оси трассы
- Iii. строение, биосинтез и биологическое действие гормонов
- Автоматизированная информационная система организации перевозок грузов по безбумажной технологии с использованием электронной накладной (АИС ЭДВ)
- Анализ устойчивости дискретных систем.
- Анатомическое строение корней. Первичная и вторичная структура корня.

Публикации по технике и механике
Публикации по биологии

Публикации по информатике

Публикации по строительству
Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по истории