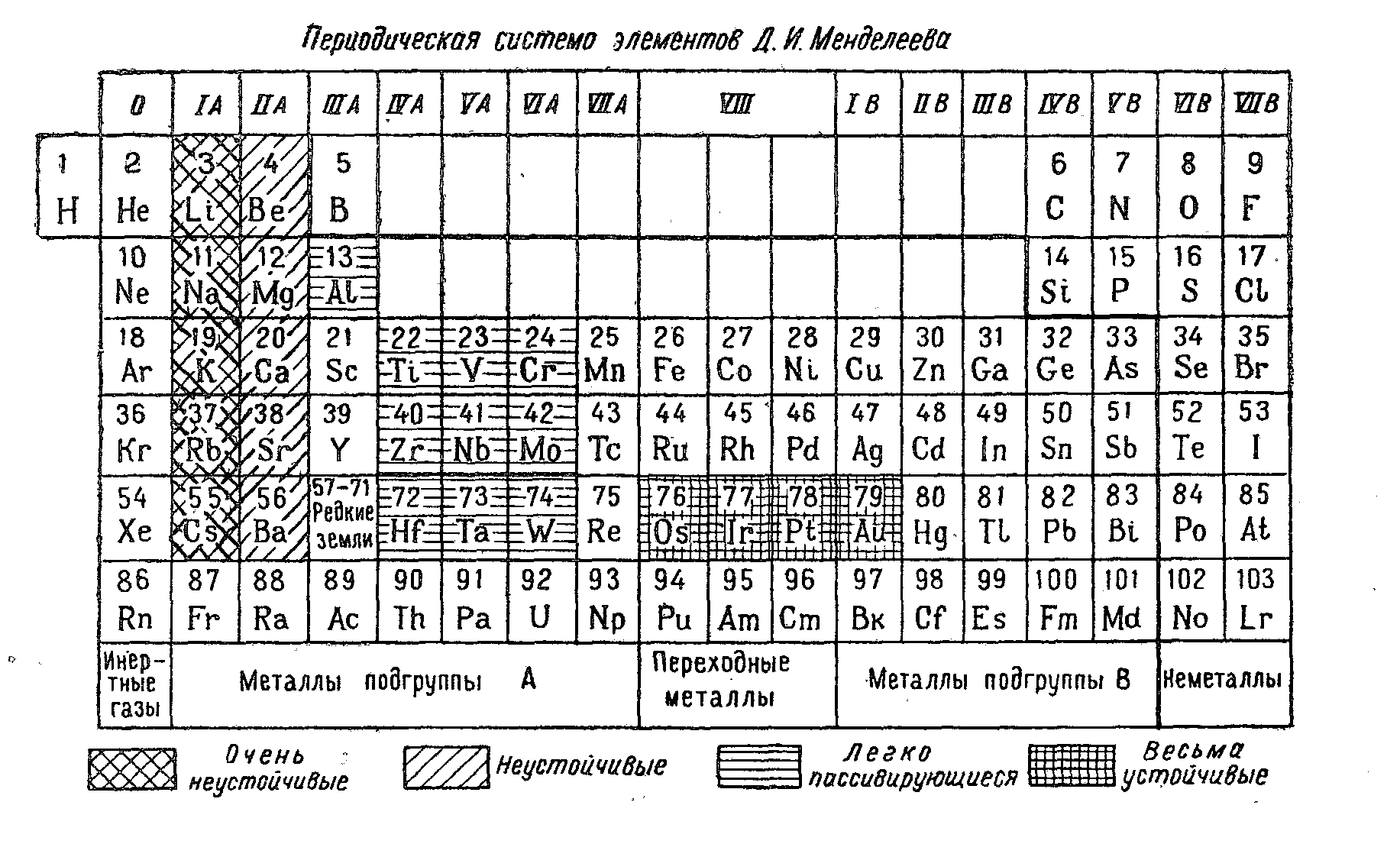
КОРРОЗИОННАЯ АКТИВНОСТЬ МЕТАЛЛА И ЕГО ПОЛОЖЕНИЕ В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА
Так как скорость электрохимической коррозии металлов является функцией многих факторов, положение металла в периодической системе элементов Д. И. Менделеева не характеризует однозначно его коррозионную стойкость; однако ряд закономерностей и периодически повторяющихся свойств можно проследить в этой системе и в отношении коррозионной характеристики металлов.
Наиболее коррозионно неустойчивые металлы находятся в подгруппах (А) I и II групп периодической системы элементов, это — щелочные и щелочноземельные металлы.
Металлы подгрупп A, начиная со второй, склонны образовывать пассивные пленки или пленки труднорастворимых вторичных продуктов коррозии, защитные свойства которых часто определяют коррозионную стойкость металлов. Способность пассивироваться у этих металлов в каждой подгруппе растет снизу вверх, т. е. с уменьшением их атомного номера.
Коррозионная стойкость металлов подгрупп В в значительной мере определяется их термодинамической устойчивостью (которая растет в каждой подгруппе сверху вниз, т. е. с увеличением их атомного номера) и реже—образующимися защитными пленками (например, АgС1, Zn(ОН)2 и Сd(ОН)2, РbSO4).
Наиболее коррозионностойкие металлы находятся внизу группы переходных элементов (Оs, Ir, Рt) и в группе IB (Аu).
1.2. ВНУТРЕННИЕ ФАКТОРЫ ЭЛЕКТРОХИМИЧЕСКОЙ КОРРОЗИИ
МЕТАЛЛОВ
Скорость и характер процесса электрохимической коррозии металла зависят от многих факторов, действующих одновременно. К внутренним факторам электрохимической коррозии металлов относятся факторы, связанные с составом, структурой, состоянием поверхности металла, напряжениями в металле и др.
1.2.1. ТЕРМОДИНАМИЧЕСКАЯ УСТОЙЧИВОСТЬ МЕТАЛЛА
Термодинамически устойчивый металл не корродирует. Для оценки возможности самопроизвольного коррозионного разрушения металла необходимо определить знак изменения изобарно-изотермического потенциала этого процесса ∆GT или сравнить значения обратимых потенциалов анодного и катодного процессов: (Eа)обр и (Eк)обр.
Соответствующие расчеты показывают, что в атмоcфере воздуха и водных растворах электролитов большенство металлов термодинамически неустойчиво. Так, если Ag, Сu, РЬ и Нg не подвержены коррозии с водородной деполяризацией, то в присутствии кислорода все они термодинамически неустойчивы, так как возможна их коррозия вследствие кислородной деполяризации.
Хотя между коррозионной стойкостью металлов, которая характеризуется скоростью протекания термодинамически возможных электрохимических коррозионных процессов, и их термодинамическими характеристиками наблюдается некоторое соответствие (щелочные и щелочноземельные металлы, например, наименее устойчивы, а благородные металлы наиболее устойчивы), однако между ними нет простой однозначной зависимости. Металл, нестойкий в одних условиях, в других условиях часто оказывается стойким. Это обусловлено тем, что протекание термодинамически возможного коррозионного процесса бывает сильно заторможено образующимися вторичными продуктами коррозии, пассивными пленками или какими-либо другими факторами. Так, термодинамически весьма неустойчивые Тi, А1 и Мg в ряде сред коррозионностойки благодаря наступлению пассивности.
1.3. ОСНОВНЫЕ ПРИЧИНЫ ПОВЫШЕННОЙ КОРРОЗИОННОЙ
СТОЙКОСТИ МЕТАЛЛОВ
Повышенная коррозионная стойкость металлов и сплавов, обусловливающая применение их в соответствующих коррозионных средах, определяется следующими причинами.
Термодинамическая устойчивость. Повышенная коррозионная стойкость ряда металлов—Аu, Рt, Ir, Рd, Аg, Сu — во многих коррозионных средах определяется их большой термодинамической устойчивостью, количественной характеристикой которой является стандартный электродный потенциал металлов.
На коррозионной стойкости этих металлов отрицательно сказывается присутствие в коррозионной среде сильных окислителей (например, НNО3+НСl), у которых V(обр) имеет очень положительные значения, и комплексообразователей (например, NН3 или СN- ), которые сильно сдвигают значение (VMe)обр. отрицательную сторону.
Возникновение пассивного состояния. Ряд термодинамически неустойчивых металлов—Ti, Аl, Сr, Ве, Мо, Мg, Ni, Со, Fе — в окислительных средах или при анодной поляризации приобретает повышенную коррозионную стойкость, обусловленную возникновением пассивного состояния, при котором тормозится протекание анодного процесса коррозии. Склонность металлов к пассивированию количественно характеризуется сдвигом потенциала коррюзии VMe в положительную сторону по сравнению с обратимым потенциалом металла (VMe)обр. в данных условиях или степенью анодного контроля Са. На коррозионной стойкости этих металлов отрицательно сказывается наличие депассивирующих факторов: присутствие в коррозионной среде активных ионов (например, С1-) или восстановителей (например, Н2, Nа2SО3 и др.), катодная поляризация и повышение температуры.
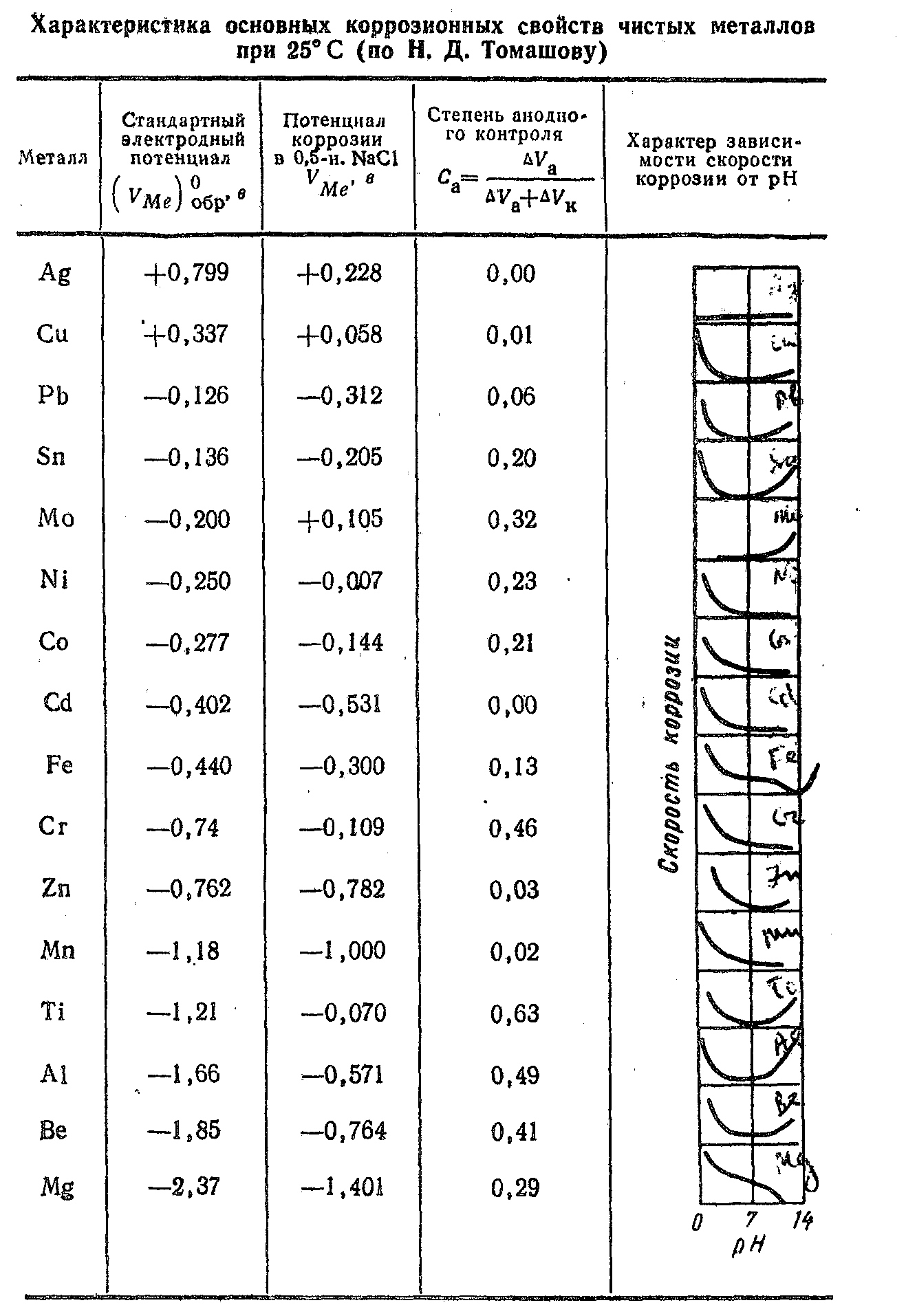
Возникновение вторичных устойчивых продуктов коррозии. Некоторые металлы в определенных средах покрываются пленками вторичных труднорастворимых продуктов коррозии, обусловливающими их повышенную коррозионную стойкость. Так, например, цинк в нейтральных растворах покрывается защитной пленкой Zn(ОН)2, свинец в растворах Н2SО4 —пленкой РbSO4, а железо в фосфатных растворах — пленкой Fе3(РO4)2.
Повышенная чистота металла. В тех случаях, когда коррозионный процесс контролируется перенапряжением водорода или перенапряжением ионизации кислорода, уменьшение содержания катодных включений за счет повышения чистоты металла увеличивает катодную поляризацию коррозионного процесса, что приводит к заметному повышению коррозионной стойкости, например чистых Zn, Fе и Аl в кислых растворах, чистого Мg в нейтральных растворах солей.
Лекция 2.
2. ГАЗОВАЯ КОРРОЗИЯ ЖЕЛЕЗА, СТАЛИ И ЧУГУНА
При высокотемпературном взаимодействии железа, стали и чугуна с воздухом, продуктами горения топлива и некоторыми другими газовыми средами имеют место различные виды газовой коррозии: окисление железа, окисление, обезуглероживание и появление водородной хрупкости стали, окисление, обезуглероживание и рост чугуна.
Процесс роста пористой (незащитной) пленки состоит из следующих отдельных стадий, протекающих последовательно:
а) переноса окислителя (например, кислорода) к поверхности раздела металл – газ;
б) адсорбции окислителя на поверхности металла;
в) реакции образования оксида.
Если образовавшийся оксид при данной температуре летуч и частично или полностью возгоняется, то имеет место еще одна стадия:
г) отвод продуктов коррозии из реакционной зоны.
Пленки, не образующие сплошного и плотного слоя (например, когда Vокс/ VМе <1), не являются защитными, так как окисляющий газ может сравнительно свободно проникать через них к поверхности металла, адсорбироваться на ней и вступать с металлом в химическую реакцию, которая является лимитирующей (наиболее заторможенной) стадией процесса. Скорость реакции в этом случае не зависит от толщины образующейся пленки и может быть выражена следующим уравнением:
dh/dτ = kс.С ,
где h – толщина образующейся пленки;
τ – время окисления металла;
kс – константа скорости химической реакции;
С– концентрация окислителя на поверхности металла, не зави-
сящая от времени вследствие очень большой легкости ад-
сорбции.
Разделив переменные, имеем
dh = kс .С.dτ .
Чтобы получить зависимость толщины пленки h от времени окисления металла τ, интегрируем предыдущее уравнение:
∫dh = kс.С.∫dτ .
Взяв неопределенный интеграл, получаем уравнение прямой (линейный закон роста пленки):
h = kс.С.τ + соnst = k1.τ + С1
где k1 = tg α (α – угол наклона прямой к оси времени);
С1= h при τ =0, т. е. такова толщина оксидной пленки на метал-
ле перед опытом.
В большинстве экспериментов значение постоянной С1 мало или равно нулю; в этом случае уравнение роста оксидной пленки принимает вид:
h = k1.τ .
Линейный закон роста оксидной пленки имеет место при окислении в воздухе и кислороде металлов, оксиды которых не удовлетворяют условию сплошности.
В защитных пленках на металлах по тем или иным причинам могут возникать следующие напряжения:
1) внутренние сжимающие напряжения, появляющиеся при росте защитной пленки, который происходит с увеличением объема, так как
Vокс/Vме>1 ;
2) внутренние напряжения сжатия на неровной поверхности металла образуют отрывающие усилия;
3) при изменении температуры в защитной пленке возникают внутренние напряжения вследствие различия коэффициентов линейного и объемного расширения металла и материала пленки, особенно заметные при резком охлаждении металла, подвергнутого газовой коррозии;
4) механические напряжения, возникающие при работе детали в конструкции (при ударах, переменных нагрузках), ухудшающие сохранность защитных пленок на металле.
2.1. ОКИСЛЕНИЕ ЖЕЛЕЗА, СТАЛИ И ЧУГУНА
Железо, углеродистая и низколегированная сталь, а также чугун при нагреве на воздухе или в продуктах горения топлива окисляются, особенно быстро при температуре выше 600° С, и покрываются продуктами газовой коррозии—окалиной.
Окисление сильно сокращает срок эксплуатации многих стальных конструкций при высоких температурах и наносит значительный ущерб металлургическому и машиностроительному производствам при горячих технологических процессах обработки металлов. Этот ущерб включает в себя:
1) угар металла (печной угар, угар при обработке металла давлением, угар при остывании металла). Потеря металла на окисление, принимая во внимание несколько нагревов, начиная от слитка до готового изделия, составляет в среднем примерно 3—3,5% от массы нагреваемого, металла;
2) разрушение огнеупорных материалов нагревательных печей окалиной с образованием сварочного шлака, что приводит к появлению бугров на подинах печей, затрудняет технологический процесс и увеличивает простои производства;
3) ускорение износа инструмента окалиной при операциях штамповки, прошивки и др.;
4) дополнительные потери металла, вызванные необходимостью удаления образовавшейся окалины с поверхности металла при его обработке давлением;
5) появление брака вследствие заковывания или закатывания окалины в металл, неравномерности закалки стали при неоднородной окалине и пр.;
6) уменьшение размеров нагреваемых деталей вследствие их окисления и др.
Все это указывает на необходимость борьбы с окислением железа, стали и чугуна при их нагреве.
2.3. СТРОЕНИЕ ОКАЛИНЫ
Железо с кислородом образует три окисла, кристаллические решетки которых приведены на рис. 1.
Закись железа FеО (вюстит) имеет простую решетку гранецентрированного куба типа каменной соли. Элементарная ячейка вюстита содержит четыре иона Fе2+ и четыре иона О2-. Этот окисел железа устойчив только при температурах выше 575°С (рис. 2). При более низких температурах он практически не образуется, а при медленном охлаждении окалины с более высоких температур распадается по уравнению
4FеО == Fе3O4 + Fе
В решетке вюстита имеются вакантные катионные места и эквивалентное количество электронных дефектов в виде ионов Fе3+ что обеспечивает хорошую ионную и электронную проводимость вюстита, который растет в результате передвижения катионов наружу по вакантным катионным местам.
Закись-окись железа Fe3O4 (магнетит), обладающая ферромагнитными свойствами, имеет сложную решетку кубической системы типа «шпинели» (рис.1). Элементарная ячейка магнетита содержит восемь ионов Fе2+, шестнадцать ионов Fе3+ и традцать два иона О2-. Fе3O4 имеет небольшой недостаток металла. Медленный рост пленки Fе3O4 во времени обусловлен слабой диффузией железа и кислорода через решетку шпинели.
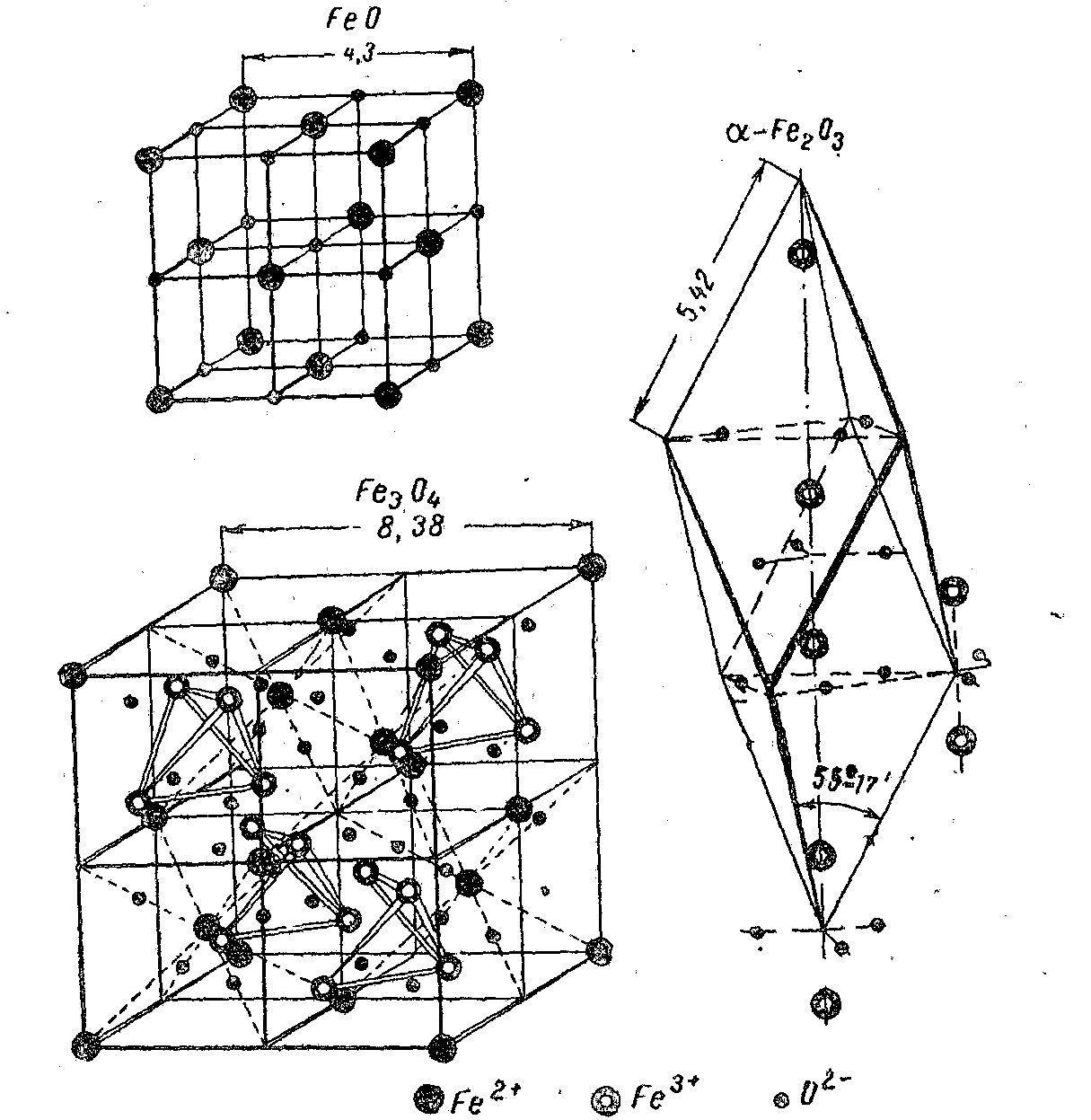
Рис. 1. Кристаллические решетки окислов железа

Рис. 2. Часть диаграммы состояния системы железо — кислород,
схема строения окалины на железе и кривая изменения концентра-
ции кислорода в слое окалины.
2.4. ОСОБЕННОСТИ КОРРОЗИИ ЖЕЛЕЗА, СТАЛИ И ЧУГУНА
Стандартный электродный потенциал железа соответствует малой его устойчивости, а по пассивируемости железо занимает среди металлов среднее положение.
На величину электродного потенциала и скорость коррозии железа, большое влияние оказывает кислород: с увеличением концентрации кислорода в растворе или облегчением его переноса к поверхности железа (например, перемешиванием электролита) потенциал последнего становится положительнее, т. е. уменьшается степень катодного контроля коррозионного процесса.
Корродируя, железо переходит в раствор в виде двухвалентных ионов:
Fе + m Н2О == Fе2+ • mН2О + 2ē,
которые при рН>5,5 образуют по труднорастворимый гидрат закиси железа белого цвета Fе(ОН)2, далее — гидрат окиси железа бурого цвета Fе(ОН)3 и при дальнейшем превращении этих продуктов—сложные гидратированные оксиды FеО • Fе2О3 • mН2О — ржавчину.
Для железа характерно длительное разблагораживание его потенциала в течение пребывания в нейтральных растворах, которое объясняется разрушением защитной окисной пленки, существовавшей на железе до его погружения в электролит, и процессом образования пленки труднорастворимых продуктов коррозии, затрудняющей доступ кислорода к поверхности корродирующего металла.
Скорость коррозии железа зависит от рН среды. В кислых растворах наблюдается равномерная коррозия железа, в нейтральных и щелочных растворах—местная (язвенная и точечная) коррозия, а в крепких растворах щелочей, особенно при повышенной температуре, — межкристаллитная коррозия.
Железо, сталь и чугун неустойчивы в кислых растворах, кроме концентрированных HNО3 (при СHNО3 от 40—50 до 94% они пассивируются), Н2SO4 (при СН2SO4 70% они покрываются нерастворимой в Н2SO4 защитной пленкой FеSO4) и их смесей (кроме чугуна, так как в этих смесях происходит своеобразное разрушение его вследствие окисления графита), Н3РO4 и ее солей [в этих растворах они покрываются защитной труднорастворимой пленкой Fе3(РO4)2] и 70—90%-ной НF (в этих растворах железо и сталь покрываются труднорастворимой пленкой.
Лекция 3.
3. ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ МЕТАЛЛОВ В ЭЛЕКТРОЛИТАХ И
МЕХАНИЗМ ИХ ВОЗНИКНОВЕНИЯ
При погружении металла в электролит в результате взаимодействия между ними возникает разность электрических потенциалов, что связано с образованием двойного электрического слоя, т. е. несимметричного распределения заряженных частиц у границы раздела фаз. Причинами возникновения этого скачка потенциала между металлом и электролитом являются:
1) переход заряженных частиц (катионов) из одной фазы в другую (из металла в электролит или из электролита в металл) с образованием двойного электрического слоя в пределах этих двух фаз (рис. 3, а и б);
2) избирательная адсорбция на поверхности металла частиц из жидкой фазы—ионов (например, С1-—рис. 3, в) или полярных молекул (например, Н2О—рис. 3, г) и появление противоположного заряда в близлежащем слое электролита с образованием двойного электрического слоя в пределах одной (жидкой) фазы;
3) образование двойного слоя, обусловленного обеими причинами, т. е. адсорбционно-ионного скачка потенциала (например, при адсорбции анионов—рис. 3, д) или при адсорбции поляризуемых молекул или атомов,
например кислорода—рис. 3, е) на поверхности металла в условиях наличия перехода катионов из металла в электролит или из электролита в металл.
Как известно из физической химии, скачок потенциала между двумя фазами не может быть измерен, но можно измерить электродвижущую силу элемента, составленного из исследуемого электрода (например, металла в электролите) и электрода, потенциал которого условно принят за нуль (стандартного водородного электрода). Эту э. д. с. принято называть потенциалом электрода, в частности электродным потенциалом металла.
Если при взаимодействии металла с электролитом (водным раствором) фазовую границу пересекают только ионы металла, то, по представлениям А. Н. Фрумкина и его школы, протекают два процесса:
1) переход этих ионов из металла в раствор с образованием, согласно Л. В. Писаржевскому, гидратированных ионов (окислительный или анодный процесс), скорость которого, измеряемая числом ионов, переходящих из фазы в фазу в единицу времени, может быть выражена через соответствующий ток ;
2) разряд этих ионов из раствора с выделением их на поверхности металла в виде нейтральных атомов, входящих в состав кристаллической решетки металла (восстановительный или катодный процесс), скорость которого также может быть выражена через соответствующий ток.
Какой из этих процессов преобладает, определяется уровнем потенциальной энергии катионов в узлах кристаллической решетки.

Рис.3. Схемы строения двойного электрического слоя.
ПОЛЯРИЗАЦИЯ И ЕЕ ВИДЫ
Причина возникновения поляризации состоит в том, что переход зарядов из металла в раствор и перемещение ионов в электролите встречает определенное сопротивление (рис.4.). В зависимости от вызывающих его факторов различают активационную, концентрационную и омическую поляризации.
Причиной активационной поляризации является сопротивление, возникающее во время катодной реакции присоединения электронов деполяризатором или торможение при переходе катионов из металлической решетки в раствор. В электрохимии эти процессы называются стадиями; считается, что самая замедленная стадия определяет скорость процесса. Преодоление такого сопротивления требует добавочной активационной энергии, поэтому и поляризация называется активационной.
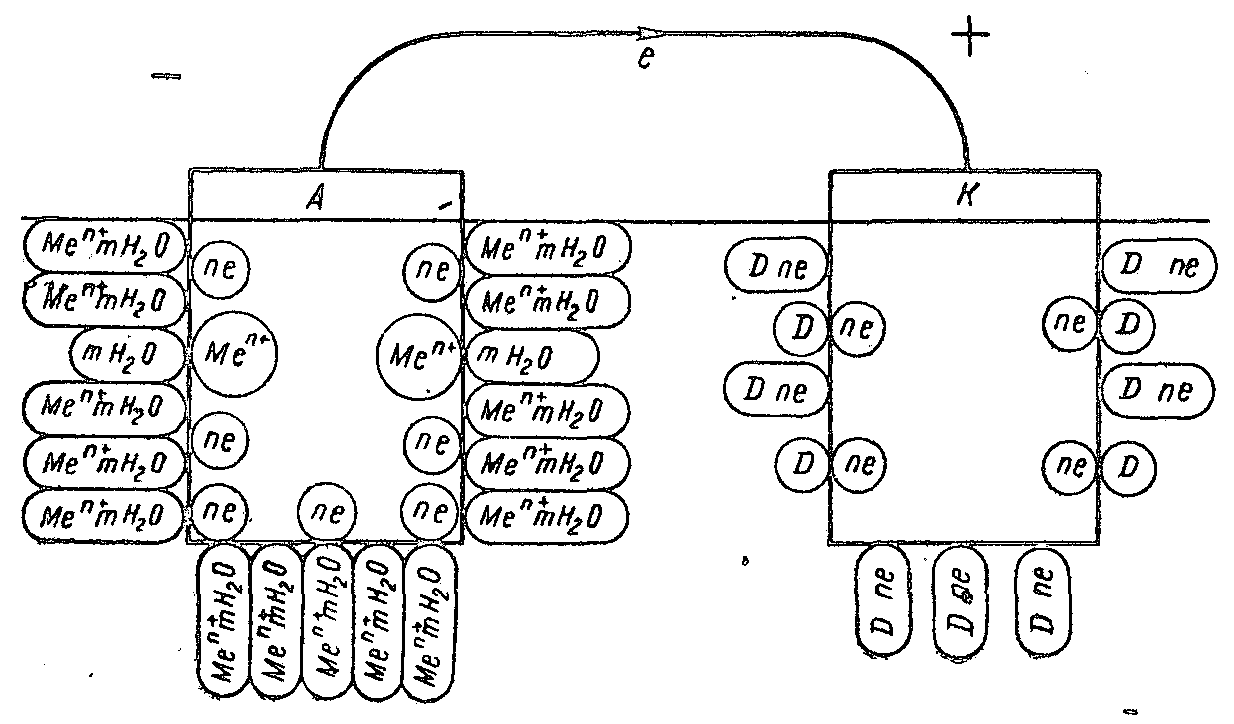
Рис.4.Схема анодной и катодной поляризации в гальваническом элементе.
При большой скорости выделения водорода в приэлектродном пространстве возникает нехватка водородных ионов. Их дальнейшее восстановление зависит от скорости их диффузии в приэлектродное пространство. Возникающая разница концентраций приводит к изменению потенциала электрода или к его поляризации. При малых скоростях электродных реакций доминирует активационная поляризация, при больших – концентрационная поляризация.
Омической поляризацией называется падение потенциала, вызываемое электрическим сопротивлением слоя электролита вблизи электрода или слоя продуктов реакции, а также обоих этих слоев одновременно. Омическая поляризация тем больше, чем выше поляризующий ток и чем больше сопротивление слоя электролита или слоя продуктов реакции. В электролитах с хорошей проводимостью, например в морской воде, величина омической поляризации очень мала, и ею практически можно пренебречь. Напротив, значительное влияние омической поляризации наблюдается в случае применения электролита с высоким сопротивлением (водопроводная вода, органические жидкости).
Активационная и омическая поляризации не зависят от перемешивания раствора, концентрационная же поляризация при перемешивании уменьшается. При правильном измерении электродного потенциала электрод сравнения или ведущий к нему электролитический ключ располагают вблизи изучаемого электрода, и омическое падение напряжения в растворе не влияет на измеренный электродный потенциал. Описываемое влияние имеет место в случае, когда в силу тех или иных практических затруднений при измерении не удается осуществить правильное расположение электродов. Поэтому в электрохимической кинетике термин «омическая поляризация электрода» не используется. В зависимости от направления сдвига потенциала электрода при прохождении постоянного тока различают анодную и катодную поляризации. Анодной поляризацией называется сдвиг потенциала в положительную сторону, катодной поляризацией – его перемещение в отрицательную сторону.
На практике всегда стремятся к увеличению поляризации в коррозионном элементе. Благодаря поляризации металлов скорость коррозии уменьшается в сотни, а то и в тысячи раз. То, что более старые конструкции корродируют медленнее, чем более новые, также является следствием поляризации. Искусственно создаваемая с помощью внешнего источника постоянного тока поляризация является основой электрохимической защиты металлов от коррозии.
| <== предыдущая лекция | | | следующая лекция ==> |
| Двоичное кодирование звука | | | ОДУВАНЧИК – СОЛНЕЧНЫЙ ЦВЕТОК |
Дата добавления: 2017-11-21; просмотров: 3114;
Поиск по сайту
Узнать еще
- A ... метка (без метки) на шатуне (стрелка) для 26.20b Измерение внутреннего диаметра
- Andantino con moto А. Бородин. Для берегов отчизны дальней
- Homo politicus и его роли
- HTML заголовок и его виды
- I-s диаграмма рабочего процесса ГТД
- I. Социально-экономическое и политическое положение Порты во второй половине XIX в.
- I. Социально-экономическое положение Ирана
- I.2. Основные категории водопотребления промышленных предприятий и их особенности

Публикации по технике и механике
Публикации по биологии

Публикации по информатике

Публикации по строительству
Публикации по физике

Публикации по химии

Публикации по электронике

Публикации по искусству

Публикации по истории